Fossils, Phylogeny, and Form:
An
Analytical Approach
by Jonathan M. Adrain, Gregory D. Edgecombe, and Bruce S. Lieberman (editors)
Topics in Geobiology vol. 19.
Kluwer Academic / Plenum Publishers, New York, 2001, 402 p.
ISBN: 0-306-46721-6. $130.00.
Morphology, Shape and Phylogeny
by Norman MacLeod and
Peter Forey (editors)
Systematics Association Special
Volume no. 64.
Taylor and Francis, London and New York, 2002, 308 p.
ISBN: 0-415-24074-3. £70.00/$125.00
|
 |

|

In the eleven years since Harvey and Pagel
(1991),
evolutionary biologists committed considerable methodological effort into the
relationship between phylogeny and numerous types of patterns. Morphology,
Shape and Phylogeny and Fossils, Phylogeny, and Form both concern
themselves with these issues. The former book deals primarily with the relationship
between phylogeny and continuous morphometric data, whereas the latter deals
with a variety of other issues also.
MacLeod and Forey's text stems from the 1999 Second
Biennial International conference of the Systematics Association with the same
name as the book. This excellent volume includes papers dealing with both aspects
of morphometric data and phylogeny: how one can infer phylogenies or even test
phylogenetic hypotheses from morphometric data, and how one can reconstruct
continuous character evolution over model phylogenies. Of course, these concepts
are inexorably linked and several of the papers treat both issues. The authors
include many of the movers and shakers of morphometric, phylogenetic and tree-based
methodology. Whether continuous data contribute to phylogenetic studies (one
of contention not long ago) receives little attention - the authors unanimously
agree that continuous characters can do so. The closest thing to a dissenting
voice is Humphries' short paper, and even he backs off from his previous stand
(e.g., Cranston and Humphries
1988) and concludes that any type of data is suitable
for phylogenetic study. Humphries does lament that ideas about transformation
and character evolution have become muddle with the concepts of sorting homologies
and classification. However, one thing made clear by many of the papers in the
volumes is that the former concept is inseparable from the two ideas. (The relationship
of classification to any of this is arbitrary philosophy, of course.)
Two distinct concepts accompany reconstructing phylogeny
from morphometric data - estimating phylogeny directly from morphometric data,
and inferring characters and character states from morphometric data. Given
the choices of qualifying continuous data or analyzing it directly, Felsenstein
opts for the latter. As he warns, it will not be easy. Of particular importance
here are the effects of correlated change. This is critical when examining multiple
landmarks associated with the same homology, but really represents an under-addressed
problem that is not unique to morphometric data (e.g., McCracken et al.
1999.)
He also briefly outlines the role of fossils in such analyses, noting two ways
in which they can be handled: using model molecular trees to estimate covariance
and rate patterns among landmarks, and then assigning fossils to their most
likely positions, or trying to simultaneously estimate (or test) morphological
parameters and phylogenetics topology. (Although Felsenstein does not mention
it, this is the only option when dealing with completely extinct taxa.) Felsenstein
also notes that the mathematical intractability of juggling so many parameters
means that we will have to use Markov Chain Monte Carlo (MCMC) methods (e.g.,
Larget and Simon 1999) to find and "integrate" over likely parameter
values. (Notably, Felsenstein does not comment on using MCMC to estimate Bayesian
probabilities of parameters.)
Other papers focus on recognizing states within
continuous characters. Rae's chapter is concerned (in large part) with the correlation
between size and shape and how that affects residuals and centroids. However,
as I note above, correlated change is a potential problem for all character
data (including molecular data). Rae advocates the use of ratios, but I am skeptical
of such approaches simply because there are numerous transformations than can
result in similar ratios among features, and because many features that one
might describe with ratios will themselves not be independently evolving ones.
Reid and Sidwell summarize methods commonly used to divide continua into discrete
characters and note that all suffer from problems. As Felsenstein notes, variation
in peak conditions among closely related species would blur boundaries even
if state transformations were fairly discrete, so perhaps these problems should
not be surprising.
Perhaps the most interesting papers in this section
are the ones dealing with geometric morphometric methods for inferring characters
and states. Chapters by Swiderski, Zelditch and Fink and by MacLeod offer interesting
points and counterpoints concerning landmark versus outline analyses. Swiderski
et al. continue earlier papers (e.g., Zelditch et al. 1995;
Swiderski et al.
1998) exploring how one might use Bookstein's thin-plate splines methods to
infer characters. In particular, they examine partial warps, which are analogous
to Factor Analysis scores, with the eigenvectors (principal warps) derived from
a description of non-uniform differences between observed forms and some reference
form. The authors repeat some of their concerns about using "traditional"
methods (e.g., principal components analysis) to identify multivariate characters
on the grounds that phylogenetic autocorrelation has strong effects on the relative
importance of eigenvectors. Also (although the authors do not state this explicitly),
phylogenetic autocorrelation will cause eigenvectors to summarize suites of
states separating clades and paraclades, and thus can miss biologically independent
features. More notably, the authors back away from some of their previous positions.
For example, they do not recommend using partial warps as a means of exploring
for characters rather than diagnosing them. They also acknowledge that outline
methods are needed to describe differences in shapes between homologous landmarks.
MacLeod illustrates some of the weaknesses of partial warps using empirical
and simulated examples. Unfortunately, the empirical trilobite example is not
a good test case - trilobite matrices (including the one used by MacLeod) have
notoriously poor resolution of states (Wagner
2000b) and the disagreement between
partial warps and nominal characters might be telling us more about the inadequacy
of the nominal characters than of partial warps. However, the inability of partial
warps to replicate known transformation patterns in simulations is damning.
MacLeod advocates using relative warps, i.e., the eigenvector summaries of multivariate
distributions among partial warps. This does separate out state types among
simulated fish nicely, especially when the analyses are confined to particular
organ systems. However, this is only a 10-taxon example and larger samples might
have relative warps confounded by phylogenetics autocorrelation. Although MacLeod
dismisses Swiderski et al.'s criticism of using multivariate summaries of distributions
to infer characters and states, eigenvectors might blend multiple warps diagnosing
particular clades, and distort warps that change occur with numerous combinations
of other warps. Similarly, eigenshape analyses (which use eigenvectors derived
from angular differences between specimen outlines and some standard outline)
run a similar risk of blurring/distorting independent derivations because phylogeny
cannot be removed. Still, eigenshape analyses are able to replicate (and improve
upon) nominal characters and states for trilobite pygidia. Given the paucity
of character states for trilobites (and, to a lesser extent, other commonly
fossilized taxa), this is at least a start. For their part, Swiderski et al.
offer measures such as smoothness, sinuosity, etc. as possible measures of outline
shapes rather than multivariate reductions such as eigenshape axes. However,
this will reintroduce the problem of categorizing continua into discrete characters
if one is to use such features as character states.
Bookstein offers a different take on identifying
character states, summarizing his creases technique. A "crease" is
the apex of a function in morphospace describing an axis of deformation between
two types and thus the transition boundary between two states. Visually, this
is the point where a D'Arcy Thomson-esque grid shows directly abutting curves
at some extrapolation angle. Although the concept is a little hairy, Bookstein
offers a simple exercise involving basic geometric features that illustrate
it nicely. Bookstein presents three empirical examples, two involving intraspecific
variation (schizophrenic vs. non-schizophrenic and male vs. female brain dimensions)
and one involving interspecific differences (hominoid species). Although Procrustes,
thin-plate splines and other methods do not clearly separate intraspecific differences,
prominent creases exist separating males from females, and schizophrenics from
non-schizophrenics. Similarly, two creases are found for hominoids (chimps vs.
hominids, and archaic vs. modern Homo sapiens) that are more prominent than
differences found by other methods. Creases obviously are very new and it is
not clear yet how important the extrapolation angles will prove to be when interpreting
these functions. Still, they do offer some hope that discrete sets do exist
in morphometric data.
Rohlf's chapter is in some ways a compliment to
Felsenstein's, with the emphasis on morphometrics and phylogeny somewhat reversed.
Rohlf notes fitting shape descriptors to phylogeny requires that factors such
as specimen orientation be factored out, something that geometric methods using
reference shapes do. However, the method used to infer ancestral conditions
also must be independent of reference shape, which is true of likelihood and
squared change parsimony, but not true of traditional linear parsimony. The
chapter initially focuses on reconstructing evolution on model trees, but it
briefly addresses inferring phylogeny from morphometric data. Rohlf briefly
suggests likelihood, squared-change parsimony or even neighbor-joining. However,
he cautions against attempting this unless many landmarks and multiple systems
are being used. This latter introduces new problems, however, as it will be
important to not allow structures with numerous landmarks to swamp the signal
of structures with few landmarks.
One concern about morphometric data is that there
is little if any phylogenetic structure present. Of course, this really should
be a concern for any character set. Cole, Lele and Richtsmeir discuss parametric
bootstrapping techniques for evaluating how well some observed measure of structure
(in this case, phenogram metrics) match real data. Parametric bootstrapping
is a cross between standard bootstrapping and typical simulations. Unlike standard
bootstrapping, which resamples from an "observed" distribution (in
this case, the data are branch lengths derived from a model tree and thus models
rather than data), a distribution is fit to the data and then used in the simulations.
Using an empirically derived tree as a model, best phenograms from simulated
data are compared to the original tree to examine how often that shape is replicated.
With structured data, this will be common. Unfortunately, the authors present
only one example using only four taxa, so the utility is difficult to assess.
Also, the approach would be most interesting if one is comparing two or more
morphometric data sets, or contrasting continuous data to discrete or molecular
data. For example, one could measure matrix structure using something like average
squared Procrustes distances and then simulate continuous evolution at different
rates (I have used similar approaches to evaluate the likelihood of amounts
of change given observed congruence and compatibility; Wagner 1998,
2001). This
then could be used to test contrast rates of change among morphometric and discrete
data sets. Also, although the authors present this as a model-tree test, they
do note that MCMC methods could be used to explore structure over a range of
trees (which in turn are weighted by the tree likelihoods) and thus make the
test of structure among continuous characters assume only that there was a phylogeny,
not a particular phylogeny.
The remaining papers focus on using model trees
to test hypotheses about continuous character evolution. These will be of especial
interest to paleontologists as the empirical examples include fossils and fossil
information about temporal branch lengths figures prominently. The latter issue
figures prominently in Polly's paper, which examines rates of rates of mammalian
tooth evolution. Two issues are important here. One is simply the length of
the branches. The second is the phylogeny used in the analysis, as there are
multiple phylogenies consistent with any given cladogram ("Eldredge's Enigma")
and these different trees will imply different rates. The importance of not
just a correct cladogram, but of a correct phylogeny (including ancestor-descendant
models) therefore is critical. Polly's analysis suggests extremely high rates
of change for mammal teeth, almost rivaling that of littorinind snail shells.
The above being said, MCMC Bayesian methods might offer a way around this, as
one could sum the probabilities of observed morphometric distributions over
multiple phylogenies (including those from different cladograms) and thus estimate
the conditional likelihoods of rates assuming only that there was an underlying
phylogeny rather than assuming a particular phylogeny.
Webster and Purvis examine the ability of several
methods to reconstruct ancestral conditions. The chapter presents a very lucid
review of what the different methods (e.g., linear parsimony, squared-change
parsimony, different likelihood methods) do and how they are related. Instead
of simulations, the authors used two model trees (a conodont family and primates)
including inferred ancestors, and asked how well inferred ancestral size matches
"observed" ancestral size. In both cases, linear parsimony actually
comes closest to matching observed ancestral sizes (based on the sum of squared
differences between observed and expected). However, the authors note that there
often were multiple equally parsimonious reconstructions, which means that the
method had multiple chances to get a correct answer. I did find it surprising
that the two-parameter likelihood model made the same reconstructions that the
one-parameter likelihood model did for the conodont data. The two-parameter
model includes both a rate parameter (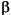 ) and a constraint parameter
(
) and a constraint parameter
(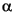 ), with
the latter affecting rates of within-lineage change relative to between lineage
change. However, I suspect that this was due to the method not taking into account
the stratigraphic ranges of the conodont species. Having a range of nearly static
values would require a high a and thus necessarily change . Also, the
authors only briefly note that the
parameters almost certainly vary across
both phylogenies. If so, then two, three, etc., b parameters increase the probability
of the observed data significantly. In addition to allowing tests for a variety
of macroevolutionary hypotheses, this should also improve the ancestral reconstructions.
), with
the latter affecting rates of within-lineage change relative to between lineage
change. However, I suspect that this was due to the method not taking into account
the stratigraphic ranges of the conodont species. Having a range of nearly static
values would require a high a and thus necessarily change . Also, the
authors only briefly note that the
parameters almost certainly vary across
both phylogenies. If so, then two, three, etc., b parameters increase the probability
of the observed data significantly. In addition to allowing tests for a variety
of macroevolutionary hypotheses, this should also improve the ancestral reconstructions.
Pagel's chapter actually addresses this and other
issues when examining hominid brain size evolution. In addition to rate and
constraint parameters, Pagel also tests for biased versus unbiased change in
brain size and shifting rates of brain size evolution. (Unfortunately, Pagel
uses different Greek letters for the parameters Webster and Purvis describe,
and and
for different parameters, which makes the first read of the paper
difficult!) Pagel finds that a positive bias parameter was significantly more
likely than an unbiased parameter, and also that an increasing rate parameter
is significantly more likely than an unchanging rate parameter. That is, brain
size tended to increase over time, and the amount of increase itself increased
over time. Pagel does make one conclusion against which I would (in general)
caution. The most likely branch scaling parameter (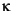 , essentially equivalent
to Webster and Purvis' a parameter) is close to 1.0, which means that the amount
of change is proportional to the branch length. Purvis asserts that if evolution
is punctuated, then
should be close to zero. However, Felsenstein notes in
his chapter that unsampled extinct ancestors invalidate this assumption. This
might not be a major problem on hominid phylogeny, which has been intensely
sampled for selfish reasons. However, in examples such as Webster and Purvis'
primate tree, the number of unsampled extinct ancestral species along branches
will be proportional to the temporal length of the branch. (This will be especially
true on longer temporal branches.) Simulations by Marcot (2000) show that this
results in punctuation and gradualism having nearly identical expectations.
, essentially equivalent
to Webster and Purvis' a parameter) is close to 1.0, which means that the amount
of change is proportional to the branch length. Purvis asserts that if evolution
is punctuated, then
should be close to zero. However, Felsenstein notes in
his chapter that unsampled extinct ancestors invalidate this assumption. This
might not be a major problem on hominid phylogeny, which has been intensely
sampled for selfish reasons. However, in examples such as Webster and Purvis'
primate tree, the number of unsampled extinct ancestral species along branches
will be proportional to the temporal length of the branch. (This will be especially
true on longer temporal branches.) Simulations by Marcot (2000) show that this
results in punctuation and gradualism having nearly identical expectations.
A question that could have made a fascinating paper
in its own right stands the original question on its head - do shape characters
have discrete states? Felsenstein suggests "no", noting that even
seemingly discrete features such as numbers of digits might represent simply
thresholds along continua. He suggests that morphometrics might allow us to
"uncode" seemingly discrete states into their constituent continuous
factors. On the other hand, Bookstein suggests "yes", that there are
discrete boundaries in morphospace (i.e., his creases). Also, the book assumes
a certain level of knowledge about morphometric techniques. However, the book
clearly is aimed at fairly advanced researchers planning to examine morphometric
data in phylogenetic contexts. Thus, the absence of such chapters in no way
detracts from what is truly excellent volume, filled with very good to excellent
(and frequently cutting edge) papers.
Fossils, Phylogeny, and Form is in some ways a more
specialized volume than is Morphology, Shape and Phylogeny in that most of the
papers deal with trilobites and in that most of the authors are trilobite specialists.
Fossils, Phylogeny, and Form also is a more conservative volume than is Morphology,
Shape and Phylogeny, being content to apply traditional analyses rather than
to push envelopes. Wills' "primer" stands out as one of the excellent
papers, one that would have made an excellent Annual Reviews paper. Although
very long, Wills deftly reviews examples of empirical morphospaces for both
discrete and morphometric data sets. He then provides a similar review of theoretical
morphospaces. In doing so, Wills provides nice descriptions of methods ranging
from simple phenetic dissimilarity to thin-plate splines. Wills does an especially
good job of describing (and illustrating) what different disparity metrics such
as variances and nearest neighbor distances tell us about morphospace, and how
contrasting metrics often is more informative than single metrics. Similarly,
Wills illustrates nicely how different morphospace patterns create different
rarefaction patterns. Finally, Wills discusses the use of morphospace studies
in morphological constraint analyses and other macroevolutionary studies, and
clearly delimits between situations where morphospaces can test hypotheses,
and where morphospace patterns only allow inferences.
Hughes and Chapman's case study with Silurian trilobites
also stands out. In particular, they wish to address the relationship between
developmental styles and phylogeny by combining Procrustes analyses and simply
phenetic analyses of trilobite populations. Hughes and Chapman report several
interesting findings showing that species are fairly discrete and that nuisance
parameters such as sampling and ontogeny contribute little to perceived variation.
One especially remarkable finding is that morphological variation does vary
in different organ systems, at least where one species is concerned. That species,
Aulacopleura konincki, shows extreme variation in thoracic segment numbers.
Although this is partly correlated, the other species are notable for there
complete lack of variation. Notably, similar types of variation are seen in
some Cambrian species. However, estimated phylogenies imply that Aulacopleura
is much more closely related to static Silurian species than to the variable
Cambrian ones. Also, thoracic variation in Aulacopleura is not accompanied by
variation elsewhere - in fact, cranidial features tend to be on the static side
(likely due to enrolling). Hughes and Chapman note that increasing the thoracic
segments increases the number of gills (and thus gill surface area) and thus
might have served as an adaptation for life in low O2 conditions. Hughes and
Chapman note that this indicates that intrinsic constraints were not inviolable
(although they are careful to note that this is very different from saying that
intrinsic constraints did not exist). An interesting follow-up to this study
would be to estimate how frequently such relaxations occurred in response to
extreme ecological environments, and whether this might have showed any temporal
trend.
The sole redundancy between Fossils, Phylogeny,
and Form and Morphology, Shape and Phylogeny exists in chapters by Zelditch,
Swiderski and Fink, and MacLeod. However, although the authors "continue"
their debates about the relative merits of different methods, both chapters
are more general than the authors' contributions in the MacLeod and Forey volume.
Thus, they should provide easier (and initially more informative) for readers
unfamiliar with morphometrics. However, readers already familiar with morphometrics
probably will get more from the authors' chapters in the MacLeod and Forey volume.
Eldredge addresses how the diagnostic characters
of higher taxa might evolve. He focuses on the expectations of the "Sloshing
Bucket" hypothesis, named after organisms' existences in two parallel biological
hierarchies (genealogical and ecological). Eldredge summarizes the expectations
of different levels on ecological processes on the derivation and fixation of
major morphological innovations. He then reviews how well trilobites fit those
expectations. Eldredge expects that ecological processes ranging from business-as-usual
to protracted long-term change all will do little to induce either speciation
or major morphological change. Eldredge expects only the highest two levels,
i.e., ecologic change being too rapid for habitat-tracking to maintain species
and (especially) full-blown mass extinctions (and their subsequent radiations)
to induce macroevolutionary patterns. Eldredge further proposes than interplay
minor turnovers (e.g., substage- or stage-level) during radiations create a
relay pattern that amplifies the affects of those radiations. Trilobites appear
to meet the expectations in some cases. Hamiltonian species ranges and Cambrian
biomeres both are consistent with pulsed turnovers and the morphologic diversification
of Ordovician trilobites is consistent with the alleged role of radiations.
However, other putative corroborating examples are problematic. The last radiation
of trilobites, the calymeniids, show the relay pattern of morphologic innovation
Eldredge predicts. However, Eldredge acknowledges that sampling of the clade
is not good and the relay pattern might reflect periodic sampling of a clade
continuously accumulating synapomorphies. Eldredge suggests that apparently
high early rates of speciation among olenelloid trilobites documented by Lieberman
(2001) support the role of radiation in clade innovation, but that analysis
probably is hindered by a highly inaccurate model phylogeny (Webster 2002 -
also, see below). Also, the relay pattern that olenelloids show could easily
be an artifact of temporally varying preservation. Thus, it is not clear that
trilobites corroborate the Sloshing Bucket hypothesis particularly well.
Ebach and Edgecombe provide an empirical review
of paleobiogeographic reconstruction. They focus on component-based methods
including Tree Mapping, Three Area Statements and Paralogy-free Subtree analysis.
Again, their examples focus on trilobites, using numerous model phylogenies
for Silurian and Devonian trilobites. Because disagreement exists concerning
the information that widespread species present for such analyses, the authors
repeat each analysis using different assumptions about those species. The results
are quite discouraging, as the methods produce very different results not only
from one another but depending on the assumptions about widespread species.
Unfortunately, these methods use phylogenetic models in lieu of data, and the
extent to which model error might affect results is not discussed. This is especially
critical when dealing with trilobites, for which minimum rates of homoplasy
are extremely high (Wagner
2000b) and often well beyond the rate at which parsimony
methods (whence the models were derived) accurately reconstruct phylogeny (see
Wagner 2000a). Thus, it is difficult to ascertain whether the lack of biogeographic
signal reflects flaws in the component-based methods or simply inaccurate assumptions
about trilobite history. Moreover, at least one of the methods (Tree Mapping)
allows only for vicariance, even though we know that dispersal is very important
in evolution, and it is questionable how well the other methods can accommodate
dispersal. However, methods that explicitly look for this pattern (e.g., Alroy
1995) are not even discussed.
What might have been more useful is a simulation
study that could examine the robustness of component-based methods to geographic
distribution assumptions and model tree error. However, approaches such as these
are rapidly becoming outdated. "Bayesian" MCMC techniques provide
tree-based tests without assuming a model tree (Huelsenbeck et al.
2000b) and
such tests have been applied to the nearly identical problem of cospeciation (Huelsenbeck et al.
2000a). With the development of likelihood tests given morphological
data (Wagner 2000c;
Lewis
2001), one could sum the likelihoods of trilobite
trees from different clades that are consistent with different biogeographic
histories (including both vicariance and dispersal). The summed likelihoods
(which approximate Bayesian posterior probabilities if we assume the prior probabilities
to be identical) then can be used to evaluate alternative biogeographic hypotheses
directly from character data rather than from intermediate phylogenetic models.
Again, it bears emphasizing that such analyses test hypotheses about geographic
dispersal assuming only that there were phylogenies, not particular phylogenies.
Lieberman applies tree-based tests of speciation
rates to Cambrian olenelloid trilobites. He emphasizes three approaches for
estimating speciation rates, Yule's pure birth (PB) model, Feller's birth-death
(BD) model, and the Bienyamé-Galton-Watson discrete time birth-death
model. Although not described as such, the first model is a special case of
the second model (i.e., pure birth is simply birth-death with an extinction
rate of zero) and the second model is a special case of the third model (where
there are "turnover" and "background" speciation rates happen
to be equal. Beginning with the pure birth model, Lieberman finds that a 90th
percentile speciation rate for the entire Phanerozoic is not particularly unlikely
for olenelloids, given a model phylogeny that suggests that olenelloids went
from 2 to 20 species in approximately six million years. Lieberman then adds
a "moderate" Phanerozoic extinction rate to that speciation rate and
finds that 2 to 20 in 6 Ma still is not improbable. From this, he concludes
that speciation rates might not have been unusually high among Cambrian trilobites.
Unfortunately, the approach taken is fundamentally flawed. What should instead
have been done is to take post-Cambrian trilobite clades as well as the Cambrian
clade and determined the likelihood of the phylogenies given the same speciation
and extinction parameters. The null hypothesis (courtesy of Ockham's Razor)
is that they are equal, as this represents a special case of the test hypotheses
(i.e., separate speciation/extinction rates for Ordovician and Cambrian clades
that happen to be equal). Unless the topologies are identical (including the
temporal lengths of branches), likelihoods will always be maximized using different
rates. Instead of using a model extinction rate for the entire Phanerozoic,
the best extinction rate could be calculated. This would seem sensible given
that extinction rates for Cambrian trilobites seem to be unusually high (Foote
1988) and also given the expected relationship between speciation and extinction
rates (e.g., Walker and Valentine
1984). However, this reveals a fundamental
shortcoming of estimating extinction rates using the methods Lieberman reviews:
pure birth hypotheses always will yield higher likelihoods than will birth-death
models when making X to Y over Z Ma comparisons whenever Y >zero. That is
because P[Y=zero | PB] = 0.0 whereas P[Y=zero | BD] > 0.0, which means that
the maximum P[Y=1 | BD] is always less than the maximum P[Y=1 | PB]. Thus, the
maximum L[PB | Y=1] will always be greater than the maximum L[Y=1 | PB]. (Modifications
to these approaches by Nee et al. [1994] do produce ML extinction rates >0,
but these rates still are unrealistically low.)
One could compensate for this by estimating the
likelihood of extinction rates given the stratigraphic ranges of species that
go extinct over an interval (Foote
1997) and incorporating that into the equations.
However, this still leaves an important biasing model: the phylogeny that is
the basis for the 2 to 20 diversification, as most of the "observations"
are range extensions (Smith
1988) implicit to the tree. Even among trilobites,
olenelloids stand out as homoplasy-ridden and lacking hierarchical structure
in character state distributions (Wagner
2000b). Simulated matrices with similar
rates of homoplasy inevitably produce highly erroneous trees and erroneous trees
are strongly biased toward exaggerating range extensions rather than underestimating
them. Notably, Webster's reanalysis of olenelloid phylogeny, cited above (using
morphometrics to weed out suites of correlated characters and populations to
identify traits varying within species) suggests that far fewer olenelloid range
extensions stretch into the early Cambrian. Thus, the "data" in this
study represent a model that appears to be highly unsound.
Even if one uses an accurate phylogeny and even
if one accounts for extinction properly, there are additional potential problems.
Contrary to common assertion (e.g., Adrain and Westrop below), the distribution
of phylogenetic range extensions depends on numerous sampling factors. For example,
parameters such as variable sampling over time systematically decrease the number
of expected range extensions (Wagner
1998). Second, it must be remembered that
speciation and extinction parameters make predictions about true richness, not
sampled richness. Even if phylogeny is accurately reconstructed, the implied
richnesses depend heavily on sampled richness, which in turn depend not simply
on sampling but also on abundance distributions (Hurlbert
1971). Richness can
be estimated only by extrapolation (e.g., Efron and Thisted
1976) whereas phylogeny
can only interpolate a minimum number of unsampled ancestors (which probably
are few relative to unsampled side branches). The most obvious solution is one
to which I already have referred. Methods developed by Foote (2001) can test
speciation, extinction and sampling rate parameters simultaneously. Modifications
to these techniques could account for abundance distributions when evaluating
sampling parameters. Now, this is not to say that phylogeny will be irrelevant
- for example, one could use general phylogenetic relationships in the manner
to test for differences among clades or over time (e.g., Magallón and
Sanderson 2001). However, even there different general phylogenies should be
used (a la Sanderson and Donoghue
1994) if only to provide sensitivity analysis.
Two chapters deal with the topic of phylogenetic
reconstruction. McLennan and Brooks contribute a remarkably antediluvian introduction
to cladistics, citing not a single methods paper published after 1985. The simplistic
summary of cladistics discusses neither the conditions under which cladograms
will replicate basic phylogenetic structure, nor the numerous simulation studies
showing the variety of circumstances sufficient to mislead cladistics, nor the
numerous empirical studies showing that different data sets evolved on the same
phylogeny produce markedly different cladograms. The methods that have largely
supplanted traditional cladistics are not even mentioned. This guide might illustrate
how phylogenetics used to be done, but one would see little resembling this
approach at modern systematics meetings.
Adrain and Westrop take issue with recent methods
designed to test phylogenetic hypotheses with morphological and (especially)
stratigraphic data. Indeed, much of the paper is an attempt to discredit my
work, which the authors (mis)characterize as "voicing as many objections
as possible" to the use of traditional parsimony. Aside from the fact that
my research program actually is about testing macroevolutionary hypotheses,
I have yet to voice a novel objection to parsimony. Most of the objections to
parsimony raised by systematists in issue after issue of Systematic Biology
predate me, never mind my research career. The initial presentation of parsimony
(Edwards and Cavalli-Sforza
1964) noted that under very particular circumstances
(i.e., such low rates of change that the probability of stasis along each branch
is nearly 1.0 and with no variation in those low rates across branches) the
most probable character matrix is one in which the shortest network among the
character states (i.e., a cladogram) matches the basic phylogenetic structure.
Edwards and Cavalli-Sforza also noted that this probably never happens in the
real world. (Edwards [1996] provides an interesting account of the early history
of parsimony and phylogenetics.)
Perhaps Adrain and Westrop's misunderstandings of
modern systematics is best summarized by their claim that parsimony is empirical
and pattern-based whereas methods such as likelihood are modeling and process-based.
This claim is fundamentally incorrect in multiple ways. First, if we restrict
ourselves to character data, then both approaches are equally empirical-the
empiricism begins and ends with the character data. This, too, is the beginning
and end of the pattern- hypotheses have likelihoods only given observed patterns,
after all. Where cladistics and likelihood differ is in the parameters used
to explain/predict the character patterns. Cladistics assumes that factors other
than cladistic topology are (for all intents and purposes) irrelevant to basic
character state distributions. Likelihood assumes that phylogeny (including
branch lengths) and rates (including relative rates of change, differences in
transition rates among states, correlated change, variation in rates across
the tree, etc.) all affect character state distributions and thus that we have
no expectations given a hypothesis about one parameter without reference to
many others. Process does not enter into the picture, except insofar that the
demonstration that two parameters are significantly better than one (say, two
rates instead of one) indicates that more than one process (whatever those might
be) were at work.
Their misunderstanding of the parameter issue becomes
important when Adrain and Westrop question the relevance of these simulations
and new methods, noting that simulations are based on the premise that parsimony
is often (or even always) wrong. However, the assumption that cladistic topology
essentially determines character state distributions is false is a necessary
conclusion of empiricism and simple deduction. That is, if cladistic topology
alone determines the shortest network among character states and assuming that
there is only one phylogeny linking a set of taxa, then we should never see
different cladograms for the same taxa given different data partitions (e.g.,
two different sets of morphology, morphology and molecules, etc.) Instead, not
only are different cladograms the norm, but radically different cladograms for
the same taxa are far from uncommon. There now are only two possible conclusions:
either the creationists are correct or the basic premise of cladistics is false.
Given the preponderance of data showing that the first idea is false, this essentially
falsifies the "cladistic topology 'determines' character state distributions"
idea and seriously diminishes the trust we can place in a method requiring that
to be true. This also falsifies Patterson's "all homologies will be synapomorphies
(congruent)" premise unless we are willing to assume that some data sets
completely lack homologies for certain portions of the tree. These two details
lead to the reason why the simulations by myself and other workers are important
- given that phylogeny certainly affects (but does not determine) character
state distributions, when does the shortest network among character states begin
to reflect factors other than cladistic topology?
Still, Adrain and Westrop state that because we cannot know the actual amount
of change and homoplasy without knowing the phylogeny (a statement that is itself
false unless every species is sampled!), simulations cannot be used to evaluate
real data sets (a la Wagner
1998). However, we do not need to know absolute
rates to evaluate data - we need to be able to measure data properties such
as minimum rates of change in real and simulated matrices. We have amply demonstrated
that simulated matrices showing the same minimum rates of change as real matrices
yield erroneous cladograms. Indeed, many empirical matrices show minimum rates
exceeding the highest minimum rates of published simulations. Adrain and Westrop's
additional claim that such assessments are somehow subjective because matrix
structure metrics are affected by how characters evolved completely misses the
point; these only demonstrate that nuisance parameters such as variable rates,
correlated change, etc., have such a strong effect on character state distributions
that we cannot evaluate phylogeny without also evaluating those parameters.
Adrain and Westrop's discussion about the utility
of stratigraphic data and how workers are using it reveals additional misconceptions
concerning phylogenetic methods. They distinguish between intrinsic information
(e.g., morphology and other things that are a heritable properties of individuals)
and extrinsic information (e.g., stratigraphic ranges and other things that
are not directly properties of individuals) and the relevance of such data to
phylogenetic issues. Because phylogeny concerns inheritance, they argue that
shared morphologies offer evidence of relationships whereas absences of stratigraphic
gaps do not. This is entirely true and it entirely misses the point. We are
(or at least I am) not interested in gathering evidence to support phylogenetic
inferences. Instead, we are interested in testing ideas about phylogeny and
other aspects of evolution with evidence. Hypothesis testing can be done only
in a deductive framework (i.e., where one explicitly states the expectations
of a hypothesis or set of hypotheses and then determines whether observations
deviate from those expectations). The only criterion for whether data of any
sort test a hypothesis is: does a hypothesis make predictions about what that
data should look like? If so, and if the observations are met, then the hypothesis
passes this test and is considered (for now) to be one of the likely explanations.
If not, the hypothesis fails the test and is considered unlikely. (Note that
Boolean modus tollens deduction is a special case of a likelihood argument,
in which the likelihood is zero and the hypothesis must be false - thus, falsification
also is a special case of a likelihood argument.) Adrain and Westrop advocate
an orthogonal logic known as abduction (see Sober
1988). Abduction is exemplified
by the saying that (given that ducks have flat bills, waddle and quack) if it
looks, walks and quacks like a duck, then it is a duck. Essentially, abduction
is unquantified Bayesian probability in which we assert that the probability
of a waddling quacking billed-thing being a duck is 1.0. Of course, without
Bayesian formalization, this "common sense" really is logical fallacy.
(For those keeping score, induction is different yet again, representing arguments
to support generalizations such as "ducks quack", which is an assumption
for either deduction or abduction.)
The likelihood:Bayesian dichotomy exemplifies the
difference in logic applied by workers such as myself and that applied by workers
such as Adrain and Westrop. If we can assume that most species sharing a trait
do so because of common ancestry, then it is probable that they are closely
related if they share traits. Thus, if two species waddle, quack and otherwise
look like ducks, they probably belong to a clade of ducks. Of course, if rates
of homoplasy are high enough, then most species sharing a trait probably do
not do so because of common ancestry, and the probability that the species share
the trait by common ancestry actually is less than the probability that they
share it because of convergence. And this is exactly why stratigraphy does not
provide information about how species are related - most contemporaneous species
in the fossil record are not close relatives. However, we expect close relatives
to be contemporaneous or nearly so. Thus, it is likely that contemporaneous
species are close relatives and unlikely that species separated by large gaps
in sampling are close relatives. It is critical to understand what I have just
written: stratigraphy provides no evidence against the idea that contemporaneous
species are relatives - the likely hypotheses pass this test. (The vast majority
of phylogenies passing a stratigraphic test will fail subsequent tests using
morphology: it is improbable that close relatives will be as different as brachiopods
and trilobites even under high rates of change, and thus phylogenies suggesting
this are unlikely.) However, the failure to sample a Silurian link between an
Ordovician and a Devonian species is unexpected if we have sampled numerous
species from the same clade and from the same environments and geographic units.
Thus, trees positing such a gap given such data fail our test and are dismissed
as unlikely. To a hypothetico-deductive worker such as myself, the concern never
is about what inferences data support unless we can deductively eliminate all
rival inferences. Instead, it is about what ideas we can reject, and which ones
require more data and better methods before we can reject them.
Of course, we need to know the expectations for
stratigraphic distributions given hypothesized durations to test hypotheses.
Adrain and Westrop's one potentially useful contribution concerns stratigraphic
sampling regimes and how environmental differences among species within the
same clade mean that we do not expect to sample members of subclade A just because
we are sampling members of subclade B. Adrain and Westrop document such environmental
heterogeneity among species within a clade of Cambrian trilobites. What would
have been interesting here is quantitative tests of species durations within
these different sampling units, using the protocol outlined by Marshall (1995;
Marshall and Ward 1996) and myself (Wagner
1995). Unfortunately, no such calculations
are made. Nor are any analyses presented that show how stratigraphic data might
have misled analyses. Indeed, a more interesting question of how changes in
environment across phylogeny affect this - if changes are common, then we actually
expect few gaps in phylogeny even if sampling only one environment simply because
lineages would keep "criss-crossing" through that environment. Conversely,
if the differences typified subclades, then we would expect simply a few long
(and not unlikely) range extensions spanning the gaps in facies sampling. Do
we need to account for more parameters when evaluating hypotheses given stratigraphic
data? Almost certainly. However, the same statement is true for morphologic
data.
All of this could be rendered moot if, as Adrain
and Westrop imply, the failure is not with parsimony but with comparative data.
Students of mammal teeth and snail shells should either get their acts together
or get different groups, they say. This is quite an audacious statement coming
from trilobite workers given that trilobite matrices often look almost as much
like the work of random number generators as of the work of Markov processes
(and almost never so well structured as most gastropod matrices, I might add!).
Moreover, advances in comparative data often add homoplasy as well as reduce
it. For example, Webster's above-cited analyses reduced homoplasy among olenelloid
trilobites substantially for many characters, but also added homoplasy for others
by showing that states linking supraspecific OTU's in early analyses varied
homoplastically among species within those OTU's. My own survey of published
studies (Wagner
2000b) found that further study added homoplasy quite frequently.
Given this, I have little hope that comparative analyses will ever become sophisticated
enough that they will reduce coded homoplasy to levels where parsimony has much
of a chance to recover phylogeny. It is not time to get new groups; instead,
it is time to adopt methods that use logical formalisms and dismiss those based
solely on philosophy.
In summary, MacLeod and Forey have produced a masterful
volume that represents an important read for workers integrating morphometric
and phylogenetic analyses in any capacity. The papers deal with a wide range
of issues pertinent to paleontologists in particular and evolutionary biologists
in general, and it offers the potential to provide both solutions to problems
and inspirations for new analyses. However, I cannot recommend Adrain et al.'s
volume. Although there are some outstanding papers, too many cling to yesterday's
methods and it offers little advancement. Readers would do better to devote
the same time to catching up on the advances in analytic methods regularly presented
in Systematic Biology, Evolution and Paleobiology.