

 A review of “pyrite disease” for paleontologists, with potential focused interventions
A review of “pyrite disease” for paleontologists, with potential focused interventions
Article number: 23(3):a44
https://doi.org/10.26879/1044
Copyright Paleontological Society, September 2020
Author biography
Plain-language and multi-lingual abstracts
PDF version
Submission: 14 November 2019. Acceptance: 31 August 2020.
ABSTRACT
A literature review of the chemical reactions involved in pyrite oxidation/hydration (“pyrite disease”) in geological or fossil collections gives direction to a suite of focused interventions. Reactions on the pyrite surface lie at the heart of the chemical process, accompanied by the movement of electrons on and through the pyrite semiconductor. Control of these electrons ultimately may be futile, but examination of the reactions at the atomic level leads to new possibilities for treatment, and identifies why some treatments are ineffective. This review also identifies gaps in our understanding of the oxidation and hydration reactions.
Fossil biomaterials–bone or shell–often contain or are partially replaced by pyrite. Acidity produced by pyrite decay destroys these materials. Older treatments focus on acid neutralization, and humidity control as solutions for megascopic problems. Focused interventions target specific steps in the chemical reactions at the atomic scale. Interventions are informed by identification of all the minerals participating in the process, and accurate characterization of the bone, pyrite and sedimentary matrix. Bone minerals and pyrite are structurally and chemically heterogeneous, so each intervention requires bench testing, complimented by analytical measurements. Chemically passivating the unreacted pyrite surface shows the greatest promise.
Anticipation of a new specimen’s susceptibility to decay is key. Decay may be accelerated using techniques of humidity buffering with saturated salt solutions. Conductivity of pyrite (which varies over four orders of magnitude) may be measured. A database of pyrite conductivity is highly desirable.
R. Chris Tacker. North Carolina Museum of Natural Sciences, 11 West Jones Street, Raleigh, North Carolina 27601-1029, USA. christopher.tacker@naturalsciences.org and Department of Marine, Earth and Atmospheric Sciences, North Carolina State University, Campus Box 8208, Raleigh, North Carolina 27695, USA. rctacker@ncsu.edu
Keywords: pyrite; oxidation; humidity; collections; treatment; fossils
Final citation: Tacker, R. Chris. 2020. A review of "pyrite disease" for paleontologists, with potential focused interventions. Palaeontologia Electronica, 23(3):a44. https://doi.org/10.26879/1044
palaeo-electronica.org/content/2020/3159-pyrite-disease
Copyright: September 2020 Paleontological Society.
This is an open access article distributed under the terms of Attribution-NonCommercial-ShareAlike 4.0 International (CC BY-NC-SA 4.0), which permits users to copy and redistribute the material in any medium or format, provided it is not used for commercial purposes and the original author and source are credited, with indications if any changes are made.
creativecommons.org/licenses/by-nc-sa/4.0/
INTRODUCTION
The late twentieth century, and early twenty-first century, saw a burst of research geared towards the understanding of the mechanisms of pyrite oxidation/hydration, which leads to acid mine drainage (AMD) through production of sulfuric acid, or its aqueous equivalent, dissolved hydrogen and sulfate ions. It was primarily driven by advances in surface science. Research was aided by the spreading availability of instrumentation such as laser Raman and Fourier Transform Infrared (FTIR) spectroscopies, as well as Secondary Electron Microscopy (SEM) and associated Energy Dispersive Spectroscopy (EDS). Further afield, the discovery of hydrous sulfate minerals on Mars led to detailed study of hydrous iron and magnesium sulfate minerals and their stability with respect to temperature and humidity.
The scientific literature on pyrite oxidation/hydration, what the curatorial community calls “pyrite disease,” is vast. The literature has never been pulled together to address the problems specific to minerals, or more properly, fossils. Pyrite adsorbs water and oxygen and grows efflorescent minerals that eventually make their own acidic puddle. Efflorescent minerals are generally hydrous ferric and ferrous iron sulfates (Howie, 1992a), but may also include alkali-bearing jarosite or aluminum-bearing alunogen or halotrichite. The higher molar volume of these minerals, compared to pyrite, mechanically wedges specimens apart as they grow (Table 1). The minerals eventually produce strong acid that is immediately destructive to fossil biomaterials, calcite, and to bone apatite.
Curators and collections staff have long noticed the problems of pyrite decaying within collections, raising concerns about protection and preservation of specimens. Many of these questions have long since been answered, but not in the curatorial literature. This paper reviews and summarizes scientific research for those who do not frequent the literature of mineralogy, of surface science, of geochemistry and environmental geochemistry, and of the Martian hydrosphere. This paper is written for the non-specialist, whether that person is an academic paleontologist, a graduate student, or an amateur. This paper is for anyone wishing to understand the problems with long-term storage of the discoveries they have made. What is required is a very basic understanding of chemistry.
The overall reaction of pyrite oxidation, hydrolysis, and dissolution is often presented as an aqueous reaction (Jambor et al., 2000):
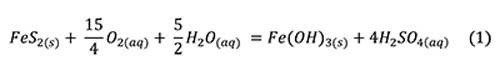
Sulfuric acid is a strong acid that dissociates nearly completely in water and is usually cited as the culprit in specimen destruction. Low pH generates Fe3+ (Jambor et al., 2000):
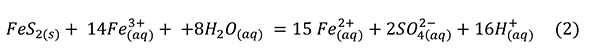
This reaction proceeds spontaneously and is exothermic enough to significantly heat experiments (Acero et al., 2007) and mine waters (Nordstrom et al., 2000).
Overall reactions are valid chemical descriptions, but they can also pass over details in reaction mechanism, for example, speciation of the iron in solution. It describes the dissolution of pyrite in water and the production of sulfuric acid and further acidity. Slightly different reactions may be written for production of other oxidized iron minerals. The reaction, as written above, pertains to an aqueous solution with dissolved oxygen. Storage problems relate to an oxygen atmosphere with water vapor (Howie, 1977a, 1977b). The two situations are fundamentally different. There is a great deal of story between reactants (“once upon a time”) and products (“the end”).
This review paper focuses solely on the abiotic reactions between pyrite, water, and oxygen. Some studies have proposed that “pyrite disease” may indeed have a bacterial component. Bacterial attack (e.g., Acidithiobacillus ferrooxidans, A. thiooxidans) on pyrite is widely studied for environmental reasons, as a significant contributor to AMD and metals release (Nordstrom et al., 2015; Blackmore et al., 2018). Bioleaching of metal sulfides has become an important process in mining (Vera et al., 2013). The scientific literature on the beneficial and detrimental effects of these bacteria is huge.
However, the importance of bacteria under dry museum storage conditions was questioned as early as 1977 (Howie, 1977a; 1977b) and has largely been discarded as a hypothesis (Newman, 1999; Fenlon and Petrera, 2019). Fenlon and Petrera (2019) cite a cutoff of 95% RH, below which the bacteria are not viable. Although temperature ranges of Acidithiobacillus species vary, they commonly have optimal pH ranges <3.5 (Falagan et al., 2019). Survival of the biofilms in collections at higher pH and drier relative humidity conditions is open to question. The present understanding of pyrite oxidation and hydration in humid air is therefore the focus of this paper, not the wet or intermittently wet conditions where these bacteria are viable.
Present treatments for pyrite degradation involve brute force tactics (Newman, 1999). Efflorescent minerals are mechanically removed (often without identification), and chemicals are added for neutralization of the acid. A coating is added to block water and oxygen. Treatments described by Birker and Kaylor (1985) involved manual cleaning, and acid neutralization in ammonia gas produced by mixing polyethylene glycol and ammonium hydroxide and suspending the specimen in the fumes. Friable specimens were then consolidated in polyvinyl acetate in toluene. Treatments described in 2019 (Hellemond, 2019) differ little from those of 34 years earlier. Hellemonde (2019) describes use of an ethanol-based solution of 2-5% ethanolamine thioglyconate, which neutralizes the acids and chelates the iron from the pyrite surface. The coating in the second case is polyvinyl acetate and acrylate, “cutting off the influence of oxygen and water.”
The shortcomings of this approach are readily apparent. There are obvious size limitations to the specimen under treatment. Birker and Kaylor (1985) make clear that the toxicity of the ethanolamine thioglyconate treatment prohibits its use under some workplace safety regulations. Baars (2019) provides anecdotal evidence that coatings are not effective to protect pyrite-bearing specimens. Howie (1992b; 1992c) observes that none of the coatings commonly in use have been demonstrated to be impermeable to oxygen or water.
Relative humidity (expressed as %RH) is the variable most amenable to control, so storage options most often turn to excluding water vapor. However, recent experiments have shown that some efflorescent minerals persist to very low humidity, along with the film of water coating the grains (Wang et al., 2013; Liu and Wang, 2015) and the pyrite grains (Jerz and Rimstidt, 2004). Low humidity does not control contributions of electrons or of water from other hydrous minerals such as clays. Finally, low humidity storage does nothing to control oxygen, the primary culprit in oxidation.
A different approach is needed. The problem can be reduced to four chemical reactions. First is the oxidation of the pyrite to ferric or ferrous sulfates. The second is growth and hydration of efflorescent minerals. Third is the production of acid from the efflorescent minerals. The final reaction is that of fossil material with strong acid. Examination of these four reactions leads to possible interventions focused on inhibiting or exploiting specific processes.
Each stage has been studied, but in separate branches of the physical sciences. Pyrite is a semiconductor of electrons, a candidate for solar energy material, and an abundant ore of iron. Efflorescent minerals, hydrous iron- and iron-aluminum sulfates, transition from subaerial to subaqueous environment and release metals into the environment as well as sulfuric acid (Jerz and Rimstidt, 2003), a process widely known as acid mine drainage (AMD). Additional interest in the hydrated sulfates grew from discoveries in the Martian soils (Farrand et al., 2009). Finally, the reaction between fossil bone—an apatite-like mineral variably altered through diagenensis—and acid has been studied as a model for bacterial decay in teeth, where acid destroys the hydroxylapatite-like enamel. Mining of phosphatic ore is followed by beneficiation with sulfuric acid to produce superphosphates and phosphoric acid. Examination of each stage in turn helps build a holistic view of the process, and points to interventions can be that most helpful.
CHEMISTRY OF THE FOUR STAGES OF PYRITE DEGRADATION
Oxidation and Hydration of Pyrite
Oxidation is an electrochemical reaction involving loss of an electron (e-). The oxidation of iron can be written as Fe2+- e- = Fe3+. The oxidizer can be any electron acceptor, another Fe3+, O2 gas, bicarbonate of soda, a co-existing mineral, or organic chemical fumes from a storage case degassing. The effects of oxygen and water are presented here, to limit the scope of the review, although the oxidizing effects of Fe3+ (Rimstidt and Vaughan, 2003) also come into play. This section benefits from the study of pyrite oxidation in surface science, environmental geochemistry, and mineralogy.
The oxidation of pyrite is reviewed in several articles (Jerz and Rimstidt, 2003; Abraitis et al., 2004; Murphy and Strongin, 2009) and excellent review volumes (Alpers et al., 2000; Vaughan, 2006). These are recommended, but pertinent parts are presented here. The literature must be treated judiciously, because most articles address conditions with dissolved oxygen in water, while our environment of interest involves subaerial conditions of abundant oxygen with variable water vapor (Howie, 1977a; 1977b). Jerz and Rimstidt (2004) demonstrated that pyrite oxidation rates are higher in air than in water, over short time scales.
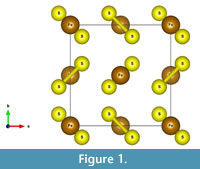 The structure of pyrite (Fe2+S2-) is shown in Figure 1, with S2 “dumbbells” alternating with iron. Oxidation is the loss of electrons, in this case the loss of an electron from Fe2+ to produce Fe3+, and the loss of seven electrons to produce S6+ from S-. Rimstidt and Vaughan (2003) note that electrons may be exchanged only in ones or twos, so that the full oxidation of the sulfur atom is likely the rate-limiting step. As a general rule in sulfides, the metal atoms provide a pathway for electron mobility, rather than the sulfur atoms (Rimstidt and Vaughan, 2003; Pearce et al., 2006).
The structure of pyrite (Fe2+S2-) is shown in Figure 1, with S2 “dumbbells” alternating with iron. Oxidation is the loss of electrons, in this case the loss of an electron from Fe2+ to produce Fe3+, and the loss of seven electrons to produce S6+ from S-. Rimstidt and Vaughan (2003) note that electrons may be exchanged only in ones or twos, so that the full oxidation of the sulfur atom is likely the rate-limiting step. As a general rule in sulfides, the metal atoms provide a pathway for electron mobility, rather than the sulfur atoms (Rimstidt and Vaughan, 2003; Pearce et al., 2006).
Studies in ultrahigh vacuum on fresh or cleaned pyrite surfaces show similar results though different spectroscopic techniques are used (X-ray photoelectron spectroscopy, XPS; scanning tunneling microscopy and spectroscopy, STM; ultraviolet photoelectron spectroscopy, UPS). The iron atom is attacked first by either O2 or H2O separately, producing Fe3+ (Guevremont et al., 1998a; 1998b; 1998c; Rosso et al., 1999; Rosso and Vaughan, 2006a; 2006b), but the effect of O2 or H2O alone is far less than both together. Rosso et al. (1999) observed that water would dissociate on the surface iron sites only if there was dissociated O2 attached at a nearby Fe3+. Rosso and Vaughan (2006) summarize the data as competitive adsorption of O2 or H2O: Oxidation of Fe2+ is accomplished by O2, which facilitates the dissociation of water on the surface. Reduced sulfur is then oxidized by charge redistribution from the oxidized iron. This scheme is supported by ab initio calculations (Stirling et al., 2003) and later work (Chandra and Gerson, 2011). Use of 18O as a tracer confirms that most sulfate oxygen or OH- radicals originate with water (Reedy et al., 1991), following the attack of oxygen on the iron.
Spectroscopic studies with synchrotron radiation using XPS in air show rapid reaction of surficial sulfur to sulfate within minutes to seconds (Schaufuss et al., 1998a; 1998b). Humidity and temperature were not controlled in these experiments. Schaufuss et al. (1998a; 1998b) showed that initial electrons are donated to the sulfur atom from iron, leading to surficial Fe3+. The proximity effect involves the surface electrons (Becker et al., 2001; Rosso and Becker, 2003), such that oxidation of Fe2+ will affect atoms nearby, explaining the patchy nature of oxidation observed. Yet the most important observation made by Schaufuss and colleagues was that the process was not simply surficial, but involved electron transfer from atoms below the surface as well. The reactions measured by Jerz and Rimstidt (2004) also indicated that subsurface atoms participate in the surficial oxidation.
The preceding research yields several important points, in summary. Iron atoms are important in initiating the oxidative reaction with O2 or H2O, and initiate oxidation of the sulfur atom by donating an electron or an OH. Iron atoms in the body and surface of the pyrite are not isolated from surface reactions, but provide electrons to participate in the reactions, as well as providing a path for diffusion of the electrons through the bulk pyrite. “Pyrite disease” does not simply proceed from the outside to the inside of the pyrite, eroding the surface to expose fresh and unaffected material. Rather, the entire body of the pyrite is involved. An external rind of oxidation on pyrite does not protect the inside from oxidation, the loss of electrons.
The factors that influence the rate of electron conduction, or charge conduction, must be examined, given the central role of electron transfer in this problem. Museum curators are well aware that all pyrites are not created equal. Some will persist for years in a pristine state, while others decay rapidly (Howie, 1992b). The answers to those questions come from Materials Science.
Pyrite is a semiconductor. The band gap in pyrite, the energy difference between electrons held in valence bands and those mobile in conduction, is small, only 0.9 eV (Abraitis et al., 2004). Conduction of charge is significantly enhanced by the presence of trace elements, leading to n-type (higher valence state, excess electrons) or p-type (lower valence state, or electron hole, or fewer electrons) semiconduction (Abraitis et al., 2004; Pearce et al., 2006). The n-type semiconductors have electrons as a majority carrier of charge. The majority carriers in p-type semiconduction are the electron holes, which essentially behave as a positively charged particle. The minority carriers in each type are electron holes in n-type and electrons in p-type. A single pyrite crystal may contain both n-type and p-type semiconductors (Rimstidt and Vaughan, 2003). Reviews of pyrite conductivity (Abraitis et al., 2004) show that it varies over four orders of magnitude, with a lengthy list of trace and minor element substitutions. Concentrations of these substitutions also cover four or more orders of magnitude (Abraitis et al., 2004). Structural and surficial defects play an additional role in the diffusion of electrons (Guevremont et al., 1998a, 1998d, 1998c; Rosso et al., 2000). Surficial sulfur deficiencies accelerate oxidation by allowing an oxygen atom to substitute directly in the sulfur vacancy (Birkholz et al., 1991; Belzile et al., 2004).
The intrinsic variability of charge conduction in pyrite provides an explanation for greatly varying rates of oxidation and hydration for multisource pyrite specimens in a collection (Savage et al., 2008). General factors affecting the reaction rate are surface area of the reactants, concentration of reactants, and temperature of the reaction (Lasaga, 1981). A small grain of pyrite will have a larger surface area, and therefore more surficial electrons that are mobile. Specific factors demonstrated to affect the reaction rate of pyrite are grain size (Lowson, 1982; Howie, 1992b), and illumination (Schoonen et al., 2000; Borda et al., 2003). The preceding review makes it clear that oxygen is an essential reactant. Atmospheric water, or relative humidity, is the other variable. Illumination, oxygen content, humidity, and temperature are amenable to control, although grain size and conductivity are not.
A megascopic view of “pyrite disease” is that humidity and acids must be controlled, to varying degrees of success. By contrast, an atomic scale perspective shows that oxygen needs control as well. If possible, surficial iron must be passivated, and likely sulfur as well. Electrons within the pyrite bulk are active participants in the oxidation process (Rimstidt and Vaughan, 2003), as are surficial electrons (Becker et al., 2001; Rosso and Becker, 2003). Realistic control of “pyrite disease” requires control of bulk and surficial electrons, which may ultimately be futile once oxidation has begun.
Growth of Efflorescent Minerals
A short list of efflorescent sulfate minerals is given in Table 1, where a clear progression in oxidation and hydration states is possible for simple ferrous and ferric sulfates. Under subaerial conditions, efflorescent mineral assemblages evolve with time, in the mine tailings pile (Jerz and Rimstidt, 2003), in collections (Blount, 1993), and in experiment (Xu et al., 2009). Yet Blount (1993) showed that the changes in hydration and mineral assemblage were sluggish and incomplete. Moreover, Xu et al. (2009) showed experimentally that the ferric sulfate minerals that form during hydration/dehydration at a given humidity depended on those initially present. When humidity is changed, the precursor minerals influence the mineral assemblage that grows. Data of Xu et al. (PO2009) strongly suggest that the reaction path - reaction kinetics - is a greater influence than equilibrium. Surface science and environmental sciences provide the descriptions of these reaction paths.
Ferrous sulfates (Fe2+SO4 • nH2O) appear to be the first to develop on the pyrite surface, mirroring the oxidation reaction. Some clues garnered from FTIR (Dunn et al., 1992; 1993) and from natural environments (Jambor et al., 2000) suggest that melanterite is the first mineral to form. It is difficult to picture melanterite (• 7H2O) forming without precursors of lower hydration states, szmolnokite (• 4H2O) and rozenite (• 1H2O), even with evidence to the contrary. Indeed, Jerz and Rimstidt (2004) showed that at humidity lower than 95%, either melanterite or szmolnokite could precipitate from the damp surfaces of pyrite. These hydrous minerals may nucleate directly on the pyrite surface or precipitate from a film of solution on the pyrite surface (Jerz and Rimstidt, 2004). The thin film of water observed by Jerz and Rimstidt (2004) was confirmed by Raman spectroscopy (Wang et al., 2012). This film is a common feature of sulfate minerals as well (Wang et al., 2013). Mineral transformations taking place in that film, or in association with that film of water, represent one of the gaps in our understanding of the process.
 In addition to the film of water forming on the pyrite, and the film of water on sulfate minerals, almost all of the sulfates listed in Table 1 are hydrous and hygroscopic, and a few are deliquescent, absorbing water until they dissolve in a solution of their own making. Hygroscopic minerals thus function as “getters” for water to contribute to the pyrite reactions. Water is gathered at the distal end of the growing crystal. The base of the crystal is rooted on the pyrite, so that a gradient of chemical potential exists, and water will diffuse to the oxidizing surface. Figure 2 shows a drop of water on the tip of a halotrichite crystal as illustration.
In addition to the film of water forming on the pyrite, and the film of water on sulfate minerals, almost all of the sulfates listed in Table 1 are hydrous and hygroscopic, and a few are deliquescent, absorbing water until they dissolve in a solution of their own making. Hygroscopic minerals thus function as “getters” for water to contribute to the pyrite reactions. Water is gathered at the distal end of the growing crystal. The base of the crystal is rooted on the pyrite, so that a gradient of chemical potential exists, and water will diffuse to the oxidizing surface. Figure 2 shows a drop of water on the tip of a halotrichite crystal as illustration.
Once the oxidation process has begun, it is largely self-sustaining. A positive feedback is established where the efflorescent minerals provide water to the pyrite surface. Further oxidation of iron in the efflorescent minerals provides an additional Fe3+ oxidizer for the pyrite (Rimstidt and Vaughan, 2003). Oxidation is further sustained by movement of electrons from the bulk of the pyrite crystal (Rimstidt and Vaughan, 2003) and from the nearby surface (Becker et al., 2001; Rosso and Becker, 2003).
Production of Acidity from Efflorescent Minerals
The release of metals and acidity from the efflorescent minerals is conveniently studied in aqueous environments, but less so in the drier environment of the tailings pile or storage cabinet. The film of water on the hydrous sulfate minerals, and on the pyrite, is very concentrated in terms of dissolved solids. Commonly used predictive models are not applicable at high ionic strengths (Plummer et al., 1988; Sippola, 2012). FREZCHEM (Marion et al., 2008; Marion et al., 2010) shows promise as an equilibrium thermodynamic model describing these reactions, but the reactions tend to be dependent on the mineral precursors (Xu et al., 2009), suggesting kinetic control.
Research reviewed above brings us to the point that melanterite has been produced from the pyrite surface, along with a film of water on the pyrite and the sulfates. Melanterite deliquesces at an activity of water equal to 0.958, 95.8% RH (Apelblat, 1993). Melanterite dissolves in water, rapidly lowering the pH (Hurowitz et al., 2009), by producing acidity through hydrolysis of iron (Frau, 2000; Hurowitz et al., 2009; Frau, 2011):
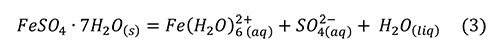
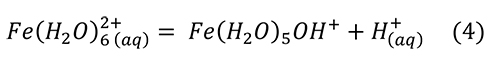
However, this reaction alone was insufficient to reproduce the pH drop observed. A much stronger acid was produced by a small amount of Fe3+ contaminant, through the reaction (Hurowitz et al., 2009; Frau, 2011).
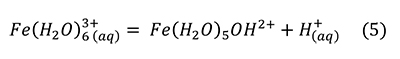
Frau (2011) verified by experiment and calculation that 0.16-0.20 wt% Fe3+ was sufficient to produce the acidity observed, that is, it was not produced by sulfuric acid.
Inferences on acidity production may be garnered through examining aqueous studies at very low pH (Nordstrom et al., 2000), and from environmental studies for predicting acidity from acid mine drainage (Li et al., 2014). At very low pH, crystallization of melanterite and coquimbite from solution have no effect on acidity production (Nordstrom et al., 2000), while rhomboclase crystallization consumes and stores hydrogen ions. Oxidized iron-bearing phases copiapite and jarosite produce 2H+ and 6H+, respectively, for each molecule of mineral precipitated (Nordstrom et al., 2000). Li et al. (2014) modelled the chemical reactions as a means of predicting acid production. Reaction of Fe3+ with water (and/or sulfate) reliably produced acidity during precipitation of amorphous Fe(OH)3, ferrihydrite, goethite, hematite, schwertmannite, and jarosite. Pyrite, melanterite, schwertmannite, and jarosite dissolve in turn, in the Li et al. (2014) model, to yield Fe(OH)3, dissolved sulfate and acidity.
Research is scarce regarding the reactions taking place in the thin film of water on the surface of the pyrite or the sulfate minerals, so extrapolation of the Nordstrom et al. (2000) and Li et al. (2014) conclusions should be done judiciously. The important result from the reactions reviewed above is that the production of acidity is not simple production of sulfuric acid, or its dissolved equivalent, except for the case of outright dissolution of pyrite in water. It is safe to assume that oxygen will continue to oxidize Fe2+ in this film of water. Precipitation of oxidized iron minerals produces acidity, as does conversion of reduced iron minerals to oxidized minerals. The presence of Fe3+ provides feedback to further oxidize the pyrite surface.
Reaction Between Bone Apatite and Acid
Sedimentary rocks that contain fossils may also contain pyrite. The fossils themselves may be partially replaced by pyrite, or contain pyrite in Haversian canals, for example. Acidity produced by pyrite oxidation and hydrolysis immediately attacks bone apatite, and immediately destroys calcite. Examination of the reaction between bone apatite and acid is the first step towards targeted interference in this reaction. The mineral apatite serves as a geochemical proxy for bone.
Valsami-Jones et al. (1998) give the following reaction for the aqueous dissolution of hydroxylapatite and fluorapatite at pH 2-7:
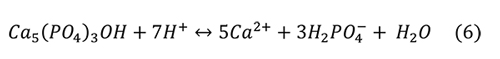
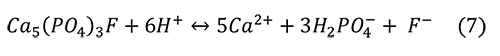
Again, the problem is that the microenvironment of interest in collections is not a solution with abundant water, but an atmosphere with humidity and a high ionic strength liquid on the surface of the pyrite or efflorescent minerals. It isn’t clear if other calcium phosphate minerals are produced from the phosphoric acid and the calcium ions, such as Ca(H2PO4)2 or CaHPO4 • H2O.
Phosphate ore is typically reacted with sulfuric acid to form gypsum and phosphoric acid (Cekinski et al., 1993b) with the overall reaction.
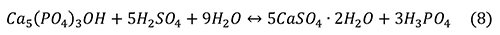
Cekinski et al. (1993b) characterized the reaction products from sulfuric acid and apatite with a variety of methods, including X-ray diffraction (XRD), X-ray photoelectron spectroscopy, and secondary electron microscopy with energy dispersive analysis (SEM-EDA). Apatite concentrates from Abraxas and Catalao, Brazil, were added to sulfuric acid and maintained at 80°C, forming a calcium sulfate (both anhydrite and minor bassanite) and a phosphate gel that coated the apatite grains. Durango fluorapatite (Young et al., 1969) reacted with sulfuric acid to form calcium sulfates and monocalcium phosphate monohydrate, Ca(H2PO4)2 • H2O. Further reaction of Ca(H2PO4)2 • H2O yielded, with time, insoluble CaHPO4 and H3PO4. If iron were added in the form Fe2O3, an additional crystalline phase was added to the previous assemblage, Fe3(H3O)H(PO4)6 • 6H2O. The addition of Fe2O3 and Al2O3 resulted in a gel-like phase mantling the apatite grains.
Further experiments elucidated the nature of the gels, using soluble iron and aluminum phosphate starting materials (Cekinski et al., 1993a) and a single Durango fluorapatite crystal rather than ore concentrates. The iron- and aluminum-free experiment gives rise to calcium sulfate and monocalcium phosphate monohydrate [Ca(H2PO4)2 • H2O]. With further hydration, calcium diphosphate (CaHPO4) forms from the monocalcium phosphate, accompanied by phosphoric acid. A gel had formed by the end of the experiment, with a composition suggested by EDA as H2SO4 • 0.5 H3PO4 • xH2O. Iron-bearing solutions of sulfuric acid produced a gel identified by XRD and EDA as Fe3(H3O)H8(PO4)6 • 6H2O. In the presence of iron, the calcium sulfates and monocalcium phosphate monohydrate were both produced much more rapidly. The experiments with both iron and aluminum produced the usual calcium sulfates and calcium phosphate, but also produced the H2SO4 • 0.5 H3PO4 • xH2O gel as well as Fe-Al-Si gels, where composition varied according to the initial amounts of the starting materials. The production of calcium sulfate was much less, due to the rapid development of the gels over the apatite.
The conclusions from this experiment are that several phosphate-bearing products are possible, including crystalline phosphates, a phosphatic gel, and phosphoric acid. The presence of iron, aluminum, and silicon likewise produces gels of a variable composition. Iron, aluminum, and silicon reactants in the experiments may be tentatively extrapolated to include iron-bearing sediments with varying amounts of aluminous clays, or quartz. Calcium sulfate minerals will reliably be a product. In my own lab, I have observed gypsum growth in the reaction between my own laboratory-grown efflorescent minerals and fossil bone. Growth of gypsum on fossils is also described by Odin et al. (2018).
On the basis of the studies presented above, one can proceed with a hypothesis for the oxidation and hydration of the pyrite and acid attack on bone apatite. The pyrite surface is rapidly attacked by oxygen and water to oxidize iron atoms, and the electrons, water and oxidants rearrange to oxidize the sulfur next. Mobile electrons on the surface and in the body of the pyrite move through networks of iron atoms and available n-type and p-type semiconductors to sustain these reactions, depending on the conductivity of the pyrite. Reduced iron sulfate minerals (Fe2+SO4 • xH2O) are released from the surface and continue to scavenge water, transferring part of it to the pyrite surface and creating a damp micro-environment. Eventually melanterite (Fe2+SO4 • 7H2O) forms, which is deliquescent (Waller, 1992). Once melanterite forms, a positive feedback loop is possible: water is further available to the pyrite surface, and oxidation of Fe2+ in melanterite to Fe3+ produces another electron acceptor. I posit the hypothesis that at this point, acidity begins to be generated in the high ionic strength liquid through oxidation and dissolution of the ferrous sulfate minerals. Bone quickly neutralizes the acids, forming gypsum or anhydrite from sulfate, along with phosphoric acid, Ca(H2PO4)2 • H2O, and amorphous phosphate minerals that have not yet been fully identified.
DISCUSSION
Each step discussed above-surface reactions, growth of efflorescent minerals, production of acidity, and attack of acid on bone-can inform a direct chemical intervention. Interventions should radically slow, if not stop, the oxidation and hydrolysis reactions. One may imagine an ideal intervention: Safety in the laboratory and collections space is paramount. The ideal intervention treatment is inexpensive, and uses readily available chemicals that may be purchased locally. Perhaps the safest chemicals are those that are approved as food additives. Application should be simple, with topical application to be preferred to immersion, but treatments should be scalable. These restrictions guide the following treatments.
The treatments described below may be described as simple chemistry, informed by experiences in the geochemical laboratory. A common thread in the curatorial literature [e.g., Howie, 1992a, or see The Geological Curator, 2019, volume 11(1)] is that historical and contemporary treatments may or may not work. This discussion offers literature-based reasons that these do not work, and suggests new interventions based on the chemical reactions reviewed. The interventions that are untried are noted as needing small-scale bench testing, not in an attempt to foist unproven remedies upon the audience, but in an attempt to involve the greater community in their development.
Interventions I - The Unaltered Pyrite
The starting point in the process of oxidation and hydration is ripe for intervention to stop, not simply forestall, deterioration. The above studies point to the central role of iron atoms in the onset and continuation of oxidation/hydration. Passivation of the surficial iron atoms is a topic of great interest in environmental geochemistry and engineering (e.g. Ouyang et al., 2015). Strategies for control of acid mine drainage are evaluated by Park et al. (2019), with advantages and disadvantages given.
Requirements for a passivating coating are that it be non-aqueous, low viscosity, and safe for use in the laboratory or collections space. Of utmost importance is its ability to bond with iron in the pyrite surface, and, ideally, with ferric or ferrous iron. The surface bonding can be demonstrated with infrared spectroscopy (either transmission or attenuated total reflectance, ATR, Li et al., 2019), a robust methodology found in nearly all chemistry departments. (Care must be taken with ATR, as sulfuric acid is the only substance that can degrade the diamond crystal.) More sophisticated spectroscopies are also available, such as confocal laser Raman or XPS.
Application of passivating chemicals is an attractive option, but more information is required about the specimen itself for the treatment to be successful, exactly as in remediation of a larger environmental site. What is the nature of porosity and cracking through the specimen (see Odin et al., 2015a)? What coatings have been previously applied? What is the percentage of pyrite throughout? These questions are answered through traditional petrography, or through high-definition CAT scanning (Larkin, 2010).
If the specimen itself is a candidate for passivation treatments, then choice of chemical is next. A short list of currently studied passivating substances shows few that are safe for use in the laboratory or the collections range. In general, a chemical that will chelate iron in nature will chelate iron in the human body, quite possibly in the lungs where heme molecules exchange oxygen. Triethylenetramine and diethylenetriamine (Chen et al., 2006), Fe3+- catecholate (Li et al., 2019), 8-hydroxyquinoline (Lan et al., 2002), and n-propyltrimethoxysilane (Diao et al., 2013) show promise for chelating iron on the pyrite surface, but are not appropriate for indoor use. Sodium acetate, various phosphate- and silicon-based chemicals (Evangelou, 1998), and linoleic acid (Wang et al., 2019) offer less toxic surface treatments for pyrite. Long-term testing is still ongoing for many of these (Camenzuli and Gore, 2013; Kang et al., 2016). No testing of these passivating substances has been conducted in the specialized context of pyrite in fossil biomaterials.
The simplest passivating substance is phosphate, delivered via solution of soluble sodium, potassium phosphate, phosphoric acid, or in bulk as commercial fertilizers (Harris and Lottermoser, 2006). The PO43- molecule reacts with Fe3+ on the pyrite surface (Evangelou, 1995; Evangelou and Zhang, 1995; Elsetinow et al., 2001). This appeared to be most successful if the surface is first treated with an oxidizing agent (Evangelou, 1995; Evangelou and Zhang, 1995; Elsetinow et al., 2001), such as hydrogen peroxide (H2O2). Later work showed that mainly Fe2+ phosphates formed, with lesser amounts of Fe3+ phosphate, iron hydroxides, and iron oxyhydroxides (Kollias et al., 2019). Kollias et al. (2019) used a solution of pH 5.5 and found a reduction in sulfur oxidation of 60%. Limits to this technique are that it is only effective at pH>3.5, and that illumination negates the effects of the phosphate in solution, allowing pyrite to react as if no phosphate were present (Elsetinow et al., 2001). The pH dependence may restrict the use in circumstances where oxidation/hydrolysis is advanced, and the effects of illumination need further examination.
Note that the idea of passivating the pyrite surface is distinct from applying various coatings like Vinac, acrylic, polyvinyl acetate, shellac, Butvar, etc. to the surface of a specimen. The rationale behind these coatings is simply to plug holes and provide a barrier. None of these have actually been demonstrated to be impervious to oxygen or water (Howie, 1992b). Data are also lacking to demonstrate that the degassing of these products does not produce oxidizers to further degrade the pyrite surface. Data are lacking to show that the coatings complex with iron or sulfur atoms on the pyrite surface. The utility of these coatings is often justified by inspection, not by any quantitative means. The long-term resistance of these coatings to microcracking is not clear. Pyrite and bone apatite expand or contract with temperature changes at different rates. Isometric pyrite expands isotropically, while apatite, a hexagonal mineral, expands anisotropically. The response of various brittle coatings to temperature changes may be cracking or crazing, so that its utility would end the first time climate control systems fail.
In the next step for protecting pyrite, minerals in specimen and matrix must be identified. What other reactions are possible? The reaction between pyrite and clay minerals has been experimentally replicated (Tacker, 2008a; 2008b; and in review), leading to abundant halotrichite and jarosite, identified by Fourier Transform Infrared Spectroscopy (FTIR) and SEM with Energy Dispersive Analysis (SEM-EDA) (Tacker, 2008a, b; and in review). The clays may participate in the reaction through electron acceptance, donation of an OH- radical, or reaction with the alumina in the clay, or as a hygroscopic mineral with adsorbed water. At present it is not clear if one mechanism predominates. Clays have been measured to rapidly transfer electrons (Hovinga et al., 2017). Oxide minerals may contain Fe3+, which is a powerful electron acceptor and oxidant. Clays and other low-temperature minerals may contain Fe3+. Sample characterization with Mossbauer spectroscopy is ideal for identifying carriers of ferric iron in a rock (Odin et al., 2015a).
Foreknowledge of the susceptibility of pyrite to oxidation is highly desirable. A glimpse of the future may be garnered through acceleration of oxidation. Humidity can be buffered at 75% with a saturated solution of sodium chloride (Greenspan, 1977) for virtually indefinite times. Samples are suspended above the solution within a sealed container. Measurement of relative humidity shows that constant humidity is established very quickly for buffers of 56% RH or higher, but less so for lower humidity (Tacker, 2008a; 2008b). Oxidation of a sample may then be examined to assist in determining the best treatments.
Susceptibility of pyrite to decay and reaction can be measured in advance. Conductivity is the most widely varying property that will influence the transmission of electrons. Conductivity is routinely measured through the Hall effect, which gives additional information about the nature of the conductor (Savage et al., 2008). There is a lower limit to sample size at present, but interesting techniques exist to overcome this. A database of conductivity measurements of museum pyrites is highly desirable and would provide a valuable predictive tool.
Finally, there is the option of actually slowing electrons with cooling. Heating tends to increase reaction rates, and cooling slows them. Data are lacking on the contraction of bone apatite and pyrite with cold, as are data for the water-bearing sulfates. The hydrous coating of iron sulfate minerals is akin to a high ionic strength liquid, very rich in solutes, which would freeze far below the usual solidus of water. Wang et al. (2013) show that the stability fields of hydrous ferric sulfates expand at lower temperatures. Higher stability at low temperatures seems to be a general feature of hydrous sulfate minerals (Wang et al., 2013). The water from these minerals would still be available for reaction with the pyrite surface, albeit at a slower rate.
Interventions II - Pyrite and Efflorescent Minerals
The most common tactic to control the growth of efflorescent minerals is to store samples at low humidity, preferably at or below 30% RH (Howie, 1992a). The goals are to limit changes in humidity and resulting changes in molar volume of efflorescent minerals (Blount, 1993), to limit further oxidation of pyrite, and to reduce the amount of water available to produce acidity. This presents a macroscopic solution to a microscopic problem. Once hygroscopic minerals have initiated, they are capable of scavenging water and transferring it to the pyrite surface, albeit at a lower rate than at elevated humidity. Humidity control does not reduce oxygen availability. Control of relative humidity likewise does nothing to reduce the movement of electrons from the bulk of the pyrite to feed oxidative reactions taking place at the surface. The equation for this reaction, properly written, describes what is happening at the atomic scale. The reaction would be between pyrite and the efflorescent minerals, which are providing water and Fe3+, not simply between the pyrite and the humidity in the air. Actual thermodynamic activity of water in the microenvironment of efflorescent minerals is higher than in the ambient atmosphere, for example, 95.8% RH in melanterite at deliquescence (Apelblat, 1993).
Storage at low humidity is inadequate to dehydrate existing ferrous sulfate minerals. Blount’s (1993) observation that hydration/dehydration reactions are sluggish has been experimentally verified. At 50°C, ambient pressure and 31.5% RH, melanterite (Fe2+SO4 • 7H2O) only dehydrates to szmolnokite (Fe2+SO4 • 1H2O) after 1000 hours (Wang and Zhou, 2014). At Mars-based pressures of 0.26-0.16 mbars and 50°C, melanterite loses water to form rozenite (Fe2+SO4 • 4H2O) and an amorphous, hydrous iron sulfate (Fe2+SO4 • xH2O) (Wang and Zhou, 2014). The higher temperature of these experiments should accelerate what would be observed at ambient temperatures (a general rule in chemical kinetics), so these reactions should take place even more slowly at 20-25°C.
Storage at low humidity is even less effective to dehydrate ferric iron sulfate minerals. Wang et al. (2012) conducted controlled humidity experiments lasting nearly four years, at 50°, 21°, and 5°C. Mineral identification was done with XRD, laser Raman, and gravimetry. The results at 31%, 11%, and 7%RH and 21°C are as follows. Ferrocopiapite [given as Fe4.67(SO4)6 (OH)2 • 20H2O] persists at these low levels of humidity for up to four years. After 30,000 hours, a compound the authors called UK#9 formed, at 11% and 7% RH. UK#9 is a hydrous ferric sulfate with unknown structure (Wang et al., 2012) but with 14-19 structural waters per formula unit (Wang et al., 2013). Rhomboclase [Fe3+H(SO4)2 • 4H2O] persists unchanged at humidity 31% and below, for four years at 21°C. Experiments with kornelite [Fe2(SO4)3 • 7H2O] at 21°C show that nothing happens to reduce the water in the formula. Wang et al. (2012) also synthesized a crystalline pentahydrated ferric sulfate (Fe2(SO4)3 • 5H2O) and an amorphous ferric sulfate of the same composition. The crystalline pentahydrate was unchanged by four years at temperatures of 50°, 21°, and 5°C and RH of 31%, 11%, and 7%. The amorphous pentahydrate persisted, but eventually crystallized rhomboclase and either kornelite or paracoquimbite [Fe2(SO4) 3 • 9H2O] at 21°C. The amorphous pentahydrate did not crystallize any minerals at higher or lower temperatures.
These experiments show that low humidity leaves water in the sulfate minerals, available to the pyrite surface for further reaction. This underscores the difference between microscopic/ atomic scale understanding of the problem, and the traditional megascopic view: actual thermodynamic activity of water in reactions with pyrite is set by water in the minerals present, not necessarily by the ambient storage atmosphere. The ferric sulfate minerals are also effective oxidizers. It is important to note that at humidity of 59% and higher, all of these minerals in the preceding paragraph (and amorphous substances) hydrated and deliquesced (Wang et al., 2012). The speed of the hydration differs in detail, but not in result.
The studies listed above support the view that the dehydration steps occur slowly or not at all. These data are at odds with research that describes the hydrous iron sulfate minerals as “metastable” (Hyde et al., 2011) and reacting rapidly. A readily testable hypothesis is that hydrous iron-bearing sulfate minerals hydrate quickly but dehydrate slowly. This hypothesis is consistent with my own observations of efflorescent mineral growth in the laboratory.
Interventions at this point often focus on the presence of a strong acid. Remedies are tailored towards neutralizing acid, but may have the unintended result of further damaging the pyrite surface. Older treatments are reviewed by Fenlon and Petrera (2019). Given the surficial reactions described above, it is clear that treatment with water-based solutions is inadvisable. Even so, I have encountered treatments with hydrogen peroxide based aqueous solutions. Hydrogen peroxide is actually used in experiments to react with the pyrite surface to accelerate oxidation (Schoonen et al., 2010). Microabrasion with carbonate minerals is sometime used for removal of efflorescent minerals, despite the fact that carbonate (Caldeira et al., 2010) and bicarbonate (Evangelou and Seta, 1997; Evangelou et al., 1998) will complex with the pyrite surface and hasten oxidation. Ammonia gas will react with sulfuric acid to form ammonium sulfate, a solid that is highly soluble in water, but not in ethanol or acetone, so removal requires water. Simple neutralization of the acid leaves the iron sulfates free to react. Chelation treatments to remove soluble iron sulfate minerals (e.g., Hellemond, 2019) leave the pyrite surface clean, but possibly pitted, ready for the next round of oxidation and hydration. Research shows that sulfur and iron atoms react with oxygen within minutes to seconds (Schaufuss et al., 1998a; 1998b) so the reactions begin anew.
A treatment that has not been bench tested is dehydration of sulfate minerals with heat under reducing atmosphere. The attraction of heating specimens to drive off water is that there is little limit to the size of specimen that can be treated in this way. Lumber companies already utilize kilns that dry wood at elevated temperatures and reducing environment. For example, the latest trend in high-end custom acoustic guitar luthiery is the use of “torrefied” tone woods, which closely reproduces the sound of much older guitars. Torrefaction kilns can attain 400°C, with controlled humidity and oxygen content. Smaller scale capabilities are already available in most experimental petrology research facilities.
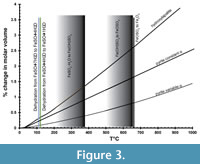 Figure 3 shows the percentage of change in molar volume of pyrite as a function of temperature (Chrystall, 1965; Fei, 1995) versus that of hydroxylapatite (Hovis et al., 2014), a common mineralogical model for bone. The thermal expansion coefficient α is given as a constant (25.7x10-6) by Fei (1995) and as a linear function of temperature (α = 8.87x10-6 + 5.82x10-9 T°C) by Chrystall (1965). Chrystall (1965) measured changes in molar volume to 400°C. Extrapolation to higher temperatures is justified by density function theory (DFT) calculations (Wen et al., 2017), which showed α as a linear function of temperatures in this range. Given that:
Figure 3 shows the percentage of change in molar volume of pyrite as a function of temperature (Chrystall, 1965; Fei, 1995) versus that of hydroxylapatite (Hovis et al., 2014), a common mineralogical model for bone. The thermal expansion coefficient α is given as a constant (25.7x10-6) by Fei (1995) and as a linear function of temperature (α = 8.87x10-6 + 5.82x10-9 T°C) by Chrystall (1965). Chrystall (1965) measured changes in molar volume to 400°C. Extrapolation to higher temperatures is justified by density function theory (DFT) calculations (Wen et al., 2017), which showed α as a linear function of temperatures in this range. Given that:
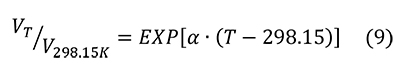
The percent change in molar volume is:
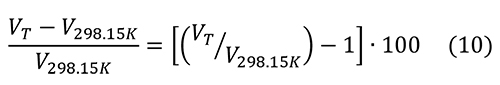
The changes in molar volume are less than 1.5% below 400°C. The thermal expansion of hydroxylapatite is slightly greater than that of pyrite, so there a possibility that heating will fracture the pyrite further.
Overlaid on thermal expansion data in Figure 3 are the data of Swami and Prasad (1980) for the dehydration of melanterite (FeSO4 • 7H2O) in air. At 110°C, melanterite dehydrates to rozenite (FeSO4 • 4H2O), then rozenite to szmolnokite (FeSO4 • H2O) at 120°C (Swami and Prasad, 1980). At higher temperatures, Fe3+(OH)SO4, then Fe3+OSO4 are gradually formed, eventually yielding hematite (Fe3O4). Data for the dehydration of different hydrous sulfates may be summarized as dehydration below 400°C, but loss of SO2 or SO3 gas above 500°C (Frost et al., 2005; Frost et al., 2006a; 2006b; Locke et al., 2007; Frost et al., 2010). Removal of water is limited to moderate temperatures below 400°C, but removal of all sulfate requires higher temperatures. What happens to pyrite at 500°C and above?
The effects of heating pyrite in various atmospheres are given in Table 2. Pyrite itself will be stable in argon atmosphere up until 500°C (Bhargava et al., 2009), at which time it breaks down into pyrrhotite (commonly written Fe1-x S where x=0-0.17) as shown by high temperature x-ray diffraction. The change to pyrrhotite structure is explained by the loss of S2 gas, leaving sulfur vacancies (Bhargava et al., 2009). Heating in N2 and CO2 atmospheres yields pyrrhotite at 600°C, with the reaction complete by 800°C (de Oliveira et al., 2018). De Oliveira et al. (2018) recommend heating in pyrite for the greatest production of pyrrhotite, excellent for their purposes but not for sample preservation. Heating in air results in evolution of SO2 gas, and above 450°C, formation of hematite (Fe2O3). The peak of reaction in argon is measured at 635°C (Thomas et al., 2003) and 665°C in nitrogen (Zhang et al., 2019). The reaction peak does not seem to shift to lower temperatures with lower pressures.
Conversion of pyrite to hematite may seem to be an attractive option that does away with pyrite entirely. In air or other atmospheres, pyrite can be left more reactive than before heating, through the presence of hematite as an oxidizer, or the presence of additional sulfur vacancies in pyrrhotite (Belzile et al., 2004). However, pyrite combusts at higher oxygen pressures. Hematite also has a larger unit cell than pyrite or marcasite, leading to potential cracking of the specimens through introduction of a larger mineral grain.
In summary, thermal dehydration is a possible treatment, but it is important that temperatures be limited to below 400°C, the thermal regime of sulfate mineral dehydration. Breakdown of the sulfate portion of the efflorescent minerals begins above 500-600°C. Breakdown of pyrite to pyrrhotite begins above 500°C. The molar volume of pyrite increases by 3% at 400°C and hydroxylapatite by about 1.5% (Figure 3). The formation of hematite is very limited at 400°C or lower. Care must be taken to dehydrate and recrystallize the sulfate minerals slowly, as rapid dehydration leaves an amorphous iron sulfate that deliquesces at lower %RH than the equivalent crystal (Sklute et al., 2015; Sklute et al., 2018).
Knowledge of the complete mineral assemblage is essential for application of thermal dehydration technique. For instance, halotrichite dehydrates by about 95% at about 340°C, but is not completely dehydrated until about 535°C (Frost et al., 2010). Bench testing on the matrix is needed to explore the possibility of changes to those minerals. Any tests for organic matter in the fossils must be conducted prior to heating.
An option that is possible for smaller specimens is dehydration of sulfate minerals under vacuum at elevated temperature. Experiments conducted at lower pressures, cited in Table 2, do not shift the loss of sulfur from pyrite to lower temperature than 500°C. The loss of water from hydrous sulfate minerals during dehydration under vacuum has not been studied, but could shift dehydration to lower temperatures.
Data reviewed above show that low-humidity storage is ineffective at reducing water in sulfate available to the pyrite surface. Given the effects of oxygen and water combined, the magnitude of the problem increases. Storage with desiccants may simply illustrate competitive scavenging for water between the efflorescent minerals and the desiccant. Data are lacking to support the hypothesis that silica or calcium sulfate is more effective than hydrous iron sulfates at attracting water.
Interventions III - Acidity
Section III, above, detailed how acidity was not produced only by the evolution of sulfuric acid. Dissolution of melanterite in its own deliquescent film of water produces acidity, but a small amount of Fe3+ greatly reduces pH (Frau, 2000; Hurowitz et al., 2009; Frau, 2011). Oxidation of ferrous hydrous sulfates produces further acidity (Nordstrom et al., 2000; Li et al., 2014). Both reactions demonstrate that control of oxygen is essential to controlling acid production.
Anoxic environments (Allington-Jones, 2019) represent the most rational approach to long-term storage of vulnerable specimens. Multilayer archival plastics are used to isolate specimens with oxygen scavengers in a low humidity environment. Bags can be customized easily for different shapes. The costs in time, money, and space are significant (Allington-Jones, 2019).
Another approach to anoxic storage would involve either vacuum storage or storage under inert gas, dry argon, or nitrogen. Dry nitrogen is used at NASA Johnson Space Center for storage of the Antarctic Meteorite Collection (Dr. Kevin Righter, curator, personal communication). Nitrogen gas (N2) will not dissociate to adsorb to the pyrite surface (Liu et al., 2012). Archival plastics mentioned above have similar order-of-magnitude diffusivities for oxygen, nitrogen, and argon (McKeen, 2017), but N2 is almost always the lowest diffusivity molecule. Bags could be purged with nitrogen gas, then inflated and sealed. The continued isolation of the specimen could be determined by inspection to determine if the bag is still full. Nitrogen generators are standard equipment in many laboratories. Purchase of a dry nitrogen generator involves up-front costs, but these costs should be compared to the long term costs of tank rental, drayage and refill.
Interventions IV - Acid and Bone
The role of bone itself in contributing to pyrite disease is probably the least studied. The surface of bone is hygroscopic. Bone apatite can carry structural OH and H2O (Yoder et al., 2012a; 2012b; Goldenberg et al., 2015) to react with pyrite. Structural water may be removed by heating above 200°C, but complete removal requires heating above 500°C (Yoder et al., 2012a). More sensitive isotopic analysis shows heating above 1500°C is required to completely remove OH (Nadeau et al., 1999). Diagenesis of bone is a highly variable process yielding a variable product (Keenan, 2016; Keenan and Engel, 2017; Kendall et al., 2018; Kontopoulos et al., 2019). Review of this subject is beyond the scope of this paper, apart from the fact that the bone itself may contain structural or adsorbed water or OH that may contribute to pyrite oxidation and hydration.
Sulfuric acid attacks the bone to produce calcium sulfate. This process may be short-circuited by flooding the specimen with a soluble calcium solution. PubChem (Kim et al., 2019) lists calcium nitrate (PubChem ID 24963) as one of the few calcium salts soluble in ethanol. Reactions of any calcium solution with sulfuric acid will liberate the two hydrogen ions to form a weaker acid or water. The simplest reaction with calcium nitrate and sulfuric acid produces calcium sulfate and nitric acid. Calcium nitrate and nitric acid are both oxidizers, so the reaction with pyrite must be considered and monitored carefully. Benefits of this reaction are that iron may be chelated with citric acid, and precipitation of calcium sulfate may serve to passivate the pyrite surface. The reaction requires bench testing and fine-tuning to explore the possibilities of competing reactions, such as that of sulfuric acid and ethanol, and the production of anhydrite or bassanite versus gypsum. The final benefit is that the molar volume of calcium sulfate is smaller than that of iron sulfates.
The phosphate molecule also acts to passivate Fe3+ on the pyrite surface, as discussed above. It is tempting to use calcium phosphate minerals or solutions to attack oxidation and sulfate production simultaneously. Few calcium phosphate minerals are water soluble, and none are alcohol soluble, so a topical application of an alcohol-based slurry may serve to address the presence of sulfate and protect the surface.
A counter-intuitive possibility is the use of aqueous solutions to treat the affected specimens. This would allow fine-tuning (concentration, pH, etc.) of the calcium salts to react with sulfuric acid. Calcium citrate tetrahydrate is sparingly soluble in water, but reacts with both sulfate and with oxidized iron. Iron (Fe3+) citrate is removed in solution and calcium sulfate precipitated. Calcium citrate is insoluble in ethanol, so subsequent washing with ethanol should remove water, and perhaps precipitate calcium citrate on pyrite as protection against future oxidation and hydrolysis. The specimen could then be dried under reducing atmosphere or vacuum depending on size. The benefit is that these reactants and products are also dietary supplements, in stark contrast to the toxicity of most iron chelating agents. This method is untested, but it gives an idea of the scope of possibilities for treatment.
The possibility of treating bone apatite to make it more resistant to acid attack is similar to fluoridating dental apatite. The list of organic acids involved in formation of dental caries includes lactic, acetic, formic, and propionic acids (Featherstone, 2008), none as strong as sulfuric acid. A fluorine-rich apatite was used in experiments (Cekinski et al., 1993b), demonstrating that simple fluoridation is ineffective against sulfuric acid attack. I have applied sulfuric acid to Durango fluorapatite to observe the reaction via FTIR microscopy, but the reaction proceeds to completion before the specimen can be placed on the microscope stage. Cekinkski et al. (1993a) noted that the development of gels during sulfuric acid attack served to protect the apatite and limit calcium sulfate formation. These gels, also noted by Blount (1993), deserve further study as a protective layer. The possibility of treating the bone to make it resistant to strong acid attack is interesting, but totally untested.
A Comment on Marcasite
Marcasite is sometimes invoked as an explanation for rapid decay of iron sulfide specimens, although the mineral identification is often unsupported. Marcasite may be readily identified in cross-polarized reflected light microscopy, as it very anisotropic, where pyrite is isotropic. Orthorhombic FeS2, marcasite, is often portrayed as “less stable” than the cubic form, pyrite. The marcasite structure is metastable with respect to the pyrite structure at higher temperature (Gronvold and Westrum, 1976; Lennie and Vaughan, 1992); this is often misinterpreted as “unstable.” Pyrite that has inverted from marcasite may be readily identified in reflected light microscopy, an important datum as it will include about two modal percent pore spaces (Murrowchick, 1992).
Quantitative measures of stability in terms of Gibbs Free Energy of Formation show small differences that nearly overlap within error (Rimstidt and Vaughan, 2003); pyrite, -160.1 ±1.7 kJmol-1 and marcasite, -158.4 ±2.1 kJmol-1 (Robie and Hemingway, 1995). The band gap for marcasite is smaller than pyrite, 0.34 eV versus 0.95 eV indicating readier electron transport (Jagadeesh and Seehra, 1980; Rimstidt and Vaughan, 2003) and perhaps quicker reaction times. Blount (1993) does not note any differences in pyrite and marcasite in a detailed study of efflorescent minerals in the collections space.
The “instability” of marcasite can be traced to the ready deterioration of “marcasite suns”, radial growths of iron sulfides in sedimentary rock from a single nucleus. This texture reflects rapid growth from a supersaturated solution (Barrie et al., 2009; Gao et al., 2016), consistent with the difficulty in nucleating iron sulfides at low temperature (Schoonen and Barnes, 1991). The crystal habit resulting from rapid growth, in turn, is rich in structural vacancies, dislocations, and small crystal domains, all of which will accelerate oxidative processes (Atanassova, 2010; Dimitrova et al., 2020).
CONCLUSIONS
Atomic-scale consideration of the reactions involved in pyrite degradation and production of acidity explain or supplant earlier observations of the problem as a megascopic scale, aqueous reaction. Electron conductivity is at the heart of the process, and may not be amenable to control. Other factors in the chemical reactions may be managed, such as oxygen availability and, classically, humidity. Passivation of the pyrite surface by stabilization of surficial iron atoms, presents a clear path forward, but is dependent on the development of non-toxic chemical treatments. Removal of water contained in efflorescent minerals by thermal dehydration is an option, but requires reducing atmosphere below 400°C. Successful treatment of specimens for “pyrite disease” will require identification of all the minerals present in the specimen. Bench testing of any treatment is essential.
The review presented herein benefits from a wide range of scientific inquiry. Advances in many of the sciences benefit the conservation of geological and fossil specimens. Perusal of the journals for conservation of art show how readily that community has embraced new microanalytical techniques. Yet the amount of targeted scientific and mineralogical study geared towards geological/fossil conservation is lacking, save for a few (Blount, 1993; Thomas et al., 2003; Odin et al., 2014, 2015a, 2015b, 2016, 2018). There are few adequate studies on museum specimens, which in themselves represent invaluable long-term experiments. Many publications simply describe the growth of hydrous sulfate minerals as “efflorescent,” without regard to the actual minerals present. Less information is available on the nature of the bone, once attacked by acid. This lack of data is not due to the negligence of museum collections professionals, but is the result of declining museum budgets, and it is a void that must be addressed.
Scientific study is lacking on the surface of the pyrite once oxidation proceeds to the point of efflorescent crystal growth. The generation of acid from the efflorescent minerals, the pyrite surface, or a film of water, is a subject ripe for examination. Passivation of the pyrite surface by chemical means needs detailed research to show that it is effective. Chemical defense of the apatite in bone with surfactants is completely unexplored, although we may be confident that fluoride-based prophylaxis of dental apatite is inadequate to protect against sulfuric acid attack.
None of these studies or interventions is inexpensive. They are costly in terms of analytical instrumentation time, in terms of staff time, in terms of space, and in terms of budget (Allington-Jones, 2019). While millions may be raised for the purchase of paleontological specimens, seldom is money included for long-term conservation research and storage. While museum budgets and staff decline, so does our ability to archive irreplaceable geological and paleontological specimens in perpetuity. As our ability to store specimens declines, so do the countless teaching and research opportunities that these specimens represent, now and in the future.
ACKNOWLEDGEMENTS
The author gratefully acknowledges the contributions of the reviewers, whose comments resulted in improvements and clarifications to the manuscript. Support for this work came from the State of North Carolina through the North Carolina Museum of Natural Sciences, and from the Friends of the Museum organization. Additional support was graciously provided by Ms. J. Edgerton, Museum Librarian. This paper would not have been possible without access to the first class research libraries at North Carolina State University.
REFERENCES
Abraitis, P.K., Pattrick, R.A.D., and Vaughan, D.J. 2004. Variations in the compositional, textural and electrical properties of natural pyrite: a review. International Journal of Mineral Processing, 74:41-59. https://doi.org/10.1016/j.minpro.2003.09.002
Acero, P., Ayora, C., and Carrera, J. 2007. Coupled thermal, hydraulic and geochemical evolution of pyritic tailings in unsaturated column experiments. Geochimica et Cosmochimica Acta, 71:5325-5338. https://doi.org/10.1016/j.gca.2007.09.007
Allington-Jones, L. 2019. Counting the costs (of low-oxygen storage projects). The Geological Curator, 11:27-31.
Alpers, C.N., Jambor, J.L., and Nordstrom, D.K. (eds.). 2000. Sulfate Minerals: Crystallography, Geochemistry, and Environmental Significance. Reviews in Mineralogy and Geochemistry, Volume 40. Mineralogical Society of America, Washington, D.C.
Apelblat, A. 1993. The vapor-pressures of saturated aqueous-solutions of potassium-bromide, ammonium-sulfate, copper(II) sulfate, iron(II) sulfate, and manganese(II) dichloride, at temperatures from 283 K to 308 K. Journal of Chemical Thermodynamics, 25:1513-1520. https://doi.org/10.1006/jcht.1993.1151
Atanassova, R. 2010. Environmental significance of pyrite with colloform textures. Comptes Rendus de L'Academie Bulgare des Sciences, 63:1335-1340.
Baars, C. 2019. Pyrite disease- into the great unknown. Geological Curator, 11:61-68.
Barrie, C.D., Boyce, A.J., Boyle, A.P., Williams, P.J., Blake, K., Ogawara, T., Akai, J., and Prior, D.J. 2009. Growth controls in colloform pyrite. American Mineralogist, 94:415-429. https://doi.org/10.2138/am.2009.3053
Becker, U., Rosso, K.M., and Hochella, M.F. 2001. The proximity effect on semiconducting mineral surfaces: A new aspect of mineral surface reactivity and surface complexation theory? Geochimica et Cosmochimica Acta, 65:2641-2649. https://doi.org/10.1016/s0016-7037(01)00624-x
Belzile, N., Chen, Y.W., Cai, M.F., and Li, Y.R. 2004. A review on pyrrhotite oxidation. Journal of Geochemical Exploration, 84:65-76. https://doi.org/10.1016/j.gexplo.2004.03.003
Bhargava, S.K., Garg, A., and Subasinghe, N.D. 2009. In situ high-temperature phase transformation studies on pyrite. Fuel, 88:988-993. https://doi.org/10.1016/j.fuel.2008.12.005
Birker, I. and Kaylor, J. 1985. Pyrite disease: case studies from the Redpath Museum. Proceedings of the 1985 Workshop on Care and Maintenance of Natural History Collections. Toronto, Ontario, Canada, p.21-27.
Birkholz, M., Fiechter, S., Hartmann, A., and Tributsch, H. 1991. Sulfur deficiency in iron pyrite (FeS2-X) and its consequences for band-structure models. Physical Review B, 43:11926-11936. https://doi.org/10.1103/PhysRevB.43.11926
Blackmore, S., Vriens, B., Sorensen, M., Power, I.M., Smith, L., Hallam, S.J., Mayer, K.U., and Beckie, R.D. 2018. Microbial and geochemical controls on waste rock weathering and drainage quality. Science of the Total Environment, 640:1004-1014. https://doi.org/10.1016/j.scitotenv.2018.05.374
Blount, A.M. 1993. Nature of the alterations which form on pyrite and marcasite during collection storage. Collection Forum, 9:1-16.
Borda, M.J., Strongin, D.R., and Schoonen, M.A.A.A. 2003. A vibrational spectroscopic study of the oxidation of pyrite by ferric iron. American Mineralogist, 88:1318-1323. https://doi.org/10.2138/am-2003-8-914
Caldeira, C.L., Ciminelli, V.S.T., and Osseo-Asare, K. 2010. The role of carbonate ions in pyrite oxidation in aqueous systems. Geochimica et Cosmochimica Acta, 74:1777-1789. https://doi.org/10.1016/j.gca.2009.12.014
Camenzuli, D. and Gore, D.B. 2013. Immobilization and encapsulation of contaminants using silica treatments: A review. Remediation: The Journal of Environmental Cleanup Costs Technologies and Techniques, 24:49-67. https://doi.org/10.1002/rem.21377
Cekinski, E., Thomassin, J.H., and Baillif, P. 1993a. Simulation of Fe-Al compounds developed during single superphosphate manufacture. Fertilizer Research, 34:259-265. https://doi.org/10.1007/bf00750572
Cekinski, E., Thomassin, J.H., and Baillif, P. 1993b. Surface investigation of the reaction products between apatite and sulfuric acid. Phosphorus, Sulfur, and Silicon, and the Related Elements, 76:495-498. https://doi.org/10.1080/10426509308032402
Chen, Y.W., Li, Y.R., Cai, M.F., Belzile, N., and Dang, Z. 2006. Preventing oxidation of iron sulfide minerals by polyethylene polyamines. Minerals Engineering, 19:19-27. https://doi.org/10.1016/j.mineng.2005.04.007
Chrystall, R.S. 1965. Thermal expansion of iron pyrites. Transactions of the Faraday Society, 61:1811-1815. https://doi.org/10.1039/tf9656101811
de Oliveira, E.M., de Oliveira, C.M., Sala, M.V.B., Montedo, O.R.K., and Peterson, M. 2018. Thermal behavior of pyrite in the CO2 and N2 atmosphere for obtaining pyrrhotite: A magnetic material. Materials Research, 21:e20170244. https://doi.org/10.1590/1980-5373-MR-2017-0244
Diao, Z.H., Shi, T.H., Wang, S.Z., Huang, X.F., Zhang, T., Tang, Y.T., Zhang, X.Y., and Qiu, R.L. 2013. Silane-based coatings on the pyrite for remediation of acid mine drainage. Water Research, 47:4391-4402. https://doi.org/10.1016/j.watres.2013.05.006
Dimitrova, D., Mladenova, V., and Hecht, L. 2020. Efforescent sulfate crystallization on fractured and polished colloform pyrite surfaces: a migration pathway of trace elements. Minerals, 10:12. https://doi.org/10.3390/min10010012
Dunn, J.G., Gong, W., and Shi, D. 1992. A Fourier transform infrared study of the oxidation of pyrite. Thermochimica Acta, 208:293-303. https://doi.org/10.1016/0040-6031(92)80173-T
Dunn, J.G., Gong, W., and Shi, D. 1993. A Fourier transform infrared study of the oxidation of pyrite. The influences of experimental variables. Thermochimica Acta, 215:247-254.
Elsetinow, A.R., Schoonen, M.A.A., and Strongin, D.R. 2001. Aqueous geochemical and surface science investigation of the effect of phosphate on pyrite oxidation. Environmental Science & Technology, 35:2252-2257. https://doi.org/10.1021/es0016809
Evangelou, V.P. 1995. Potential microencapsulation of pyrite by artificial inducement of ferric phosphate coatings. Journal of Environmental Quality, 24:535-542. https://doi.org/10.2134/jeq1995.00472425002400030021x
Evangelou, V.P. and Zhang, Y.L. 1995. A review - Pyrite oxidation mechanisms and acid mine drainage prevention. Critical Reviews in Environmental Science and Technology, 25:141-199. https://doi.org/10.1080/10643389509388477
Evangelou, V.P. and Seta, A.K. 1997. Infrared spectroscopic and wet chemistry evidence on HCO3 catalyzing pyrite oxidation. Abstracts of Papers of the American Chemical Society, 213:144-GEOC.
Evangelou, V.P. 1998. Pyrite oxidation control, p. 419-421. In Geller W., Klapper H., and Salomons, W. (eds.). Acidic Mining Lakes: Acid Mine Drainage, Limnology and Reclamation. Springer, New York. https://doi.org/10.1007/978-3-642-71954-7_23
Evangelou, V.P., Seta, A.K., and Holt, A. 1998. Potential role of bicarbonate during pyrite oxidation. Environmental Science & Technology, 32:2084-2091. https://doi.org/10.1021/es970829m
Falagan, C., Moya-Beltran, A., Castro, M., Quatrini, R., and Johnson, D.B. 2019. Acidithiobacillus sulfuriphilus sp. nov.: an extremely acidophilic sulfur-oxidizing chemolithotroph isolated from a neutral pH environment. International Journal of Systematic and Evolutionary Microbiology, 69:2907-2913. https://doi.org/10.1099/ijsem.0.003576
Farrand, W.H., Glotch, T.D., Rice, J.W., Jr., Hurowitz, J.A., and Swayze, G.A. 2009. Discovery of jarosite within the Mawrth Vallis region of Mars: Implications for the geologic history of the region. Icarus, 204:478-488. https://doi.org/10.1016/j.icarus.2009.07.014
Featherstone, J.D.B. 2008. Dental caries: a dynamic disease process. Australian Dental Journal, 53:286-291. https://doi.org/10.1111/j.1834-7819.2008.00064.x
Fei, Y. 1995. Thermal expansion, p. 29-44. In Ahrens, T. (ed.), Mineral Physics and Crystallography: A Handbook of Physical Constants. American Geophysical Union, United States of America. https://doi.org/10.1029/rf002p0029
Fenlon, A. and Petrera, L. 2019. Pyrite oxidation: a history of treatments at the Natural History Museum, London. The Geological Curator, 11:9-18.
Frau, F. 2000. The formation-dissolution-precipitation cycle of melanterite at the abandoned pyrite mine of Genna Luas in Sardinia, Italy: environmental implications. Mineralogical Magazine, 64:995-1006. https://doi.org/10.1180/002646100550001
Frau, F. 2011. Acid production by FeSO4 · nH2O dissolution: comment. American Mineralogist, 96:444-446. https://doi.org/10.2138/am.2011.3573
Frost, R.L., Weier, M.L., and Martens, W. 2005. Thermal decomposition of jarosites of potassium, sodium and lead. Journal of Thermal Analysis and Calorimetry, 82:115-118. https://doi.org/10.1007/s10973-005-0850-z
Frost, R.L., Wills, R.A., Kloprogge, J.T., and Martens, W. 2006a. Thermal decomposition of ammonium jarosite (NH4)Fe-3(SO4)2(OH)6. Journal of Thermal Analysis and Calorimetry, 84:489-496. https://doi.org/10.1007/s10973-005-6953-8
Frost, R.L., Wills, R.A., Kloprogge, J.T., and Martens, W.N. 2006b. Thermal decomposition of hydronium jarosite (H3O)Fe-3(SO4)2(OH) 6. Journal of Thermal Analysis and Calorimetry, 83:213-218. https://doi.org/10.1007/s10973-005-6908-0
Frost, R.L., Palmer, S.J., Kristof, J., and Horvath, E. 2010. Dynamic and controlled rate thermal analysis of halotrichite. Journal of Thermal Analysis and Calorimetry, 99:501-507. https://doi.org/10.1007/s10973-009-0275-1
Gao, S., Huang, F., Wang, Y.H., and Gao, W.Y. 2016. A review of research progress in the genesis of colloform pyrite and its environmental indications. Acta Geologica Sinica-English Edition, 90:1353-1369. https://doi.org/10.1111/1755-6724.12774
Goldenberg, J.E., Wilt, Z., Schermerhorn, D.V., Pasteris, J.D., and Yoder, C.H. 2015. Structural effects on incorporated water in carbonated apatites. American Mineralogist, 100:274-280. https://doi.org/10.2138/am-2015-5025
Greenspan, L. 1977. Humidity fixed-points of binary saturated aqueous solutions. Journal of Research of the National Bureau of Standards, Section A-Physics and Chemistry, 81:89-96. https://doi.org/10.6028/jres.081A.011
Gronvold, F. and Westrum, E.F. 1976. Heat-capacities of iron disulfides thermodynamics of marcasite from 5 TO 700 K, pyrite from 300 TO 780 K, and transformation of marcasite to pyrite. Journal of Chemical Thermodynamics, 8:1039-1048. https://doi.org/10.1016/0021-9614(76)90135-x
Guevremont, J.M., Bebie, J., Elsetinow, A.R., Strongin, D.R., and Schoonen, M.A.A. 1998a. Reactivity of the (100) plane of pyrite in oxidizing gaseous and aqueous environments: Effects of surface imperfections. Environmental Science & Technology, 32:3743-3748. https://doi.org/10.1021/es980298h
Guevremont, J.M., Elsetinow, A.R., Strongin, D.R., Bebie, J., and Schoonen, M.A.A. 1998b. Structure sensitivity of pyrite oxidation: comparison of the (100) and (111) planes. American Mineralogist, 83:1353-1356. https://doi.org/10.2138/am-1998-11-1225
Guevremont, J.M., Strongin, D.R., and Schoonen, M.A.A. 1998c. Thermal chemistry of H2S and H2O on the (100) plane of pyrite: unique reactivity of defect sites. American Mineralogist, 83:1246-1255. https://doi.org/10.2138/am-1998-11-1213
Harris, D.L. and Lottermoser, B.G. 2006. Evaluation of phosphate fertilizers for ameliorating acid mine waste. Applied Geochemistry, 21:1216-1225. https://doi.org/10.1016/j.apgeochem.2006.03.009
Hellemond, A. 2019. The Dendermonde Mammoth: fighting pyrite decay and the preservation of unique paleontological heritage. The Geological Curator, 11:55-59.
Hovinga, A.L., Sander, M., Bruggeman, C., and Behrends, T. 2017. Redox properties of clay-rich sediments as assessed by mediated electrochemical analysis: separating pyrite, siderite and structural Fe in clay minerals. Chemical Geology, 457:149-161. https://doi.org/10.1016/j.chemgeo.2017.03.022
Hovis, G.L., Scott, B.T., Altomare, C.M., Leaman, A.R., Morris, M.D., Tomaino, G.P., and McCubbin, F.M. 2014. Thermal expansion of fluorapatite-hydroxylapatite crystalline solutions. American Mineralogist, 99:2171-2175. https://doi.org/10.2138/am-2014-4914
Howie, F.M.P. 1977a. Pyrite and conservation Part 1: historical aspects. Newsletter of the Geological Curators' Group, 1:457-465.
Howie, F.M.P. 1977b. Pyrite and conservation: Part 2. Newsletter of the Geological Curator’s, 1:497-512.
Howie, F.M.P. 1992a. The Care and Conservation of Geological Materials: Minerals, Rocks, Meteorites and Lunar Finds. Butterworth-Heinemann, Oxford, England. https://doi.org/10.4324/9781315042626
Howie, F.M.P. 1992b. Pyrite and marcasite, p. 70-84. In Howie, F. (ed.), The Care and Conservation of Geological Materials: Minerals, Rocks, Meteorites and Lunar Finds. Butterworth-Heinemann, Oxford, U.K. https://doi.org/10.4324/9781315042626
Howie, F.M.P. 1992c. Sulphides and allied minerals in collections, p. 56-69. In Howie, F. (ed.), The Care and Conservation of Geological Materials: Minerals, Rocks, Meteorites and Lunar Finds. Butterworth-Heinemann, Oxford, U.K. https://doi.org/10.4324/9781315042626
Hurowitz, J.A., Tosca, N.J., and Dyar, M.D. 2009. Acid production by FeSO4 · nH2O dissolution and implications for terrestrial and martian aquatic systems. American Mineralogist, 94:409-414. https://doi.org/10.2138/am.2009.3085
Hyde, B.C., King, P.L., Dyar, M.D., Spilde, M.N., and Ali, A.M.S. 2011. Methods to analyze metastable and microparticulate hydrated and hydrous iron sulfate minerals. American Mineralogist, 96:1856-1869. https://doi.org/10.2138/am.2011.3792
Jagadeesh, M.S. and Seehra, M.S. 1980. Electrical resistivity and band gap of marcasite (FeS2). Physics Letters A, 80:59-61. https://doi.org/10.1016/0375-9601(80)90454-5
Jambor, J.I., Nordstrom, D.K., and Alpers, C.N. 2000. Metal-sulfate salts from sulfide mineral oxidation, p. 303-350. In Alpers, C.N., Jambor, J.L. and Nordstrom, D.K. (eds.), Sulfate Minerals - Crystallography, Geochemistry and Environmental Significance. Mineralogical Society of America, Washington, D.C. https://doi.org/10.2138/rmg.2000.40.6
Jerz, J.K. and Rimstidt, J.D. 2003. Efflorescent iron sulfate minerals: Paragenesis, relative stability, and environmental impact. American Mineralogist, 88:1919-1932. https://doi.org/10.2138/am-2003-11-1235
Jerz, J.K. and Rimstidt, J.D. 2004. Pyrite oxidation in moist air. Geochimica et Cosmochimica Acta, 68:701-714. https://doi.org/10.1016/s0016-7037(03)00499-x
Kang, C.U., Jeon, B.H., Park, S.S., Kang, J.S., Kim, K.H., Kim, D.K., Choi, U.K., and Kim, S.J. 2016. Inhibition of pyrite oxidation by surface coating: a long-term field study. Environmental Geochemistry and Health, 38:1137-1146. https://doi.org/10.1007/s10653-015-9778-9
Keenan, S.W. 2016. From bone to fossil: a review of the diagenesis of bioapatite. American Mineralogist, 101:1943-1951. https://doi.org/10.2138/am-2016-5737
Keenan, S.W. and Engel, A.S. 2017. Reconstructing diagenetic conditions of bone at the Gray Fossil Site, Tennessee, USA. Palaeogeography Palaeoclimatology Palaeoecology, 471:48-57. https://doi.org/10.1016/j.palaeo.2017.01.037
Kendall, C., Eriksen, A.M.H., Kontopoulos, I., Collins, M.J., and Turner-Walker, G. 2018. Diagenesis of archaeological bone and tooth. Palaeogeography Palaeoclimatology Palaeoecology, 491:21-37. https://doi.org/10.1016/j.palaeo.2017.11.041
Kim, S., Chen, J., Cheng, T., Gindulyte, A., He, J., He, S., Li, Q., Shoemaker, B.A., Thiessen, P.A., Yu, B., Zaslavsky, L., Zhang, J., and Bolton, E.E. 2019. PubChem 2019 update: improved access to chemical data. Nucleic Acids Research, 47:D1102-1109. https://doi.org/10.1093/nar/gky1033
Kollias, K., Mylona, E., Adam, K., Chrysochoou, M., Papassiopi, N., and Xenidis, A. 2019. Characterization of phosphate coating formed on pyrite surface to prevent oxidation. Applied Geochemistry, 110:104435. https://doi.org/10.1016/j.apgeochem.2019.104435
Kontopoulos, I., Penkman, K., McAllister, G.D., Lynnerup, N., Damgaard, P.B., Hansen, H.B., Allentoft, M.E., and Collins, M.J. 2019. Petrous bone diagenesis: a multi-analytical approach. Palaeogeography Palaeoclimatology Palaeoecology, 518:143-154. https://doi.org/10.1016/j.palaeo.2019.01.005
Lan, Y., Huang, X., and Deng, B. 2002. Suppression of pyrite oxidation by 8-hydroxyquinoline. Archives of Environmental Contamination and Toxicology, 43:168-174. https://doi.org/10.1007/s00244-002-1178-3
Larkin, N.R. 2010. Literally a ‘mammoth task’: the conservation, preparation and curation of the West Runton Mammoth skeleton. Quaternary International, 228:233-240. https://doi.org/10.1016/j.quaint.2010.07.002
Lasaga, A.C. 1981. Rate laws of chemical reactions, p. 1-68. In Lasaga, A.C. and Kirkpatrick, R.J. (eds.), Kinetics of Geochemical Processes, Mineralogical Society of America, Washington, D.C. https://doi.org/10.1515/9781501508233
Lennie, A.R. and Vaughan, D.J. 1992. Kinetics of the marcasite-pyrite transformation - An infrared spectroscopic study. American Mineralogist, 77:1166-1171.
Li, J., Kawashima, N., Fan, R., Schumann, R.C., Gerson, A.R., and Smart, R.S. 2014. Method for distinctive estimation of stored acidity forms in acid mine wastes. Environmental Science & Technology, 48:11445-11452. https://doi.org/10.1021/es502482m
Li, X., Hiroyoshi, N., Tabelin, C.B., Naruwa, K., Harada, C., and Ito, M. 2019. Suppressive effects of ferric-catecholate complexes on pyrite oxidation. Chemosphere, 214:70-78. https://doi.org/10.1016/j.chemosphere.2018.09.086
Liu, T., Temprano, I., Jenkins, S.J., D.A., K., and Driver, S.M. 2012. Nitrogen adsorption and desorption at iron pyrite FeS2 (100) surfaces. Physical Chemistry Chemical Physics, 14:11491-11499. https://doi.org/10.1039/c2cp41549f
Liu, Y. and Wang, A. 2015. Dehydration of Na-jarosite, ferricopiapite, and rhomboclase at temperatures of 50 and 95 degrees C: implications for Martian ferric sulfates. Journal of Raman Spectroscopy, 46:493-500. https://doi.org/10.1002/jrs.4655
Locke, A.J., Martens, W.N., and Frost, R.L. 2007. Thermal analysis of halotrichites. Thermochimica Acta, 459:64-72. https://doi.org/10.1016/j.tca.2007.04.015
Lowson, R.T. 1982. Aqueous oxidation of pyrite by molecular oxygen. Chemical Reviews, 82:461-497. https://doi.org/10.1021/cr00051a001
Marion, G.M., Kargel, J.S., and Catling, D.C. 2008. Modeling ferrous-ferric iron chemistry with application to martian surface geochemistry. Geochimica et Cosmochimica Acta, 72:242-266. https://doi.org/10.1016/j.gca.2007.10.012
Marion, G.M., Mironenko, M.V., and Roberts, M.W. 2010. FREZCHEM: a geochemical model for cold aqueous solutions. Computers & Geosciences, 36:10-15. https://doi.org/10.1016/j.cageo.2009.06.004
McKeen, L.W. 2017. Permeability Properties of Plastics and Elastomers (Fourth Edition). William Andrew Publishing. https://doi.org/10.1016/B978-0-323-50859-9.00018-X
Murphy, R. and Strongin, D.R. 2009. Surface reactivity of pyrite and related sulfides. Surface Science Reports, 64:1-45. https://doi.org/10.1016/j.surfrep.2008.09.002
Murrowchick, J.B. 1992. Marcasite inversion and the petrographic determination of pyrite ancestry. Economic Geology, 87:1141-1152. https://doi.org/10.2113/gsecongeo.87.4.1141
Nadeau, S.L., Epstein, S., and Stolper, E. 1999. Hydrogen and carbon abundances and isotopic ratios in apatite from alkaline intrusive complexes, with a focus on carbonatites. Geochimica et Cosmochimica Acta, 63:1837-1851. https://doi.org/10.1016/s0016-7037(99)00057-5
Newman, A. 1999. Pyrite oxidation and museum collections: a review of theory and conservation treatments. The Geological Curator, 6:363-371.
Nordstrom, D.K., Alpers, C.N., Ptacek, C.J., and Blowes, D.W. 2000. Negative pH and extremely acidic mine waters from Iron Mountain, California. Environmental Science & Technology, 34:254-258. https://doi.org/10.1021/es990646v
Nordstrom, D.K., Blowes, D.W., and Ptacek, C.J. 2015. Hydrogeochemistry and microbiology of mine drainage: an update. Applied Geochemistry, 57:3-16.
Odin, G.P., Vanmeert, F., Janssens, K., Lelievre, H., Mertz, J.D., and Rouchon, V. 2014. Accelerated ageing of shales of palaeontological interest: impact of temperature conditions. Annales De Paleontologie, 100:137-149. https://doi.org/10.1016/j.annpal.2013.12.002
Odin, G.P., Cabaret, T., Mertz, J.D., Menendez, B., Etienne, L., Wattiaux, A., and Rouchon, V. 2015a. Alteration of fossil-bearing shale (Autun Basin, France; Permian), part I: Characterizing iron speciation and its vulnerability to weathering by combined use of Mossbauer spectroscopy, X-ray diffraction, porosimetry and permeability measurements. Annales De Paleontologie, 101:75-85. https://doi.org/10.1016/j.annpal.2015.01.002
Odin, G.P., Vanmeert, F., Farges, F., Gand, G., Janssens, K., Romero-Sarmiento, M.F., Steyer, J.S., Vantelon, D., and Rouchon, V. 2015b. Alteration of fossil-bearing shale (Autun, France; Permian), part II: monitoring artificial and natural ageing by combined use of S and Ca K-edge XANES analysis, Rock-Eval pyrolysis and FTIR analysis. Annales De Paleontologie, 101:225-239. https://doi.org/10.1016/j.annpal.2015.03.001
Odin, G.P., Belhadj, O., Cabaret, T., Foy, E., and Rouchon, V. 2016. Alterations of fossil-bearing shale (Autun, France; Permian), part III: framboidal pyrite and sulfur as the main cause of efflorescence. Annales De Paleontologie, 102:31-40. https://doi.org/10.1016/j.annpal.2016.01.001
Odin, G.P., Rouchon, V., Bethoux, O., and Ren, D. 2018. Gypsum growth induced by pyrite oxidation jeopardises the conservation of fossil specimens: an example from the Xiaheyan entomofauna (Late Carboniferous, China). Palaeogeography Palaeoclimatology Palaeoecology, 507:15-29. https://doi.org/10.1016/j.palaeo.2018.05.035
Ouyang, Y., Liu, Y., Zhu, R., Ge, F., Xu, T., Luo, Z., and Liang, L. 2015. Pyrite oxidation inhibition by organosilane coatings for acid mine drainage control. Minerals Engineering, 72:57-64. https://doi.org/10.1016/j.mineng.2014.12.020
Park, I., Tabelin, C.B., Jeon, S., Li, X.L., Seno, K., Ito, M., and Hiroyoshi, N. 2019. A review of recent strategies for acid mine drainage prevention and mine tailings recycling. Chemosphere, 219:588-606. https://doi.org/10.1016/j.chemosphere.2018.11.053
Pearce, C.I., Pattrick, R.A.D., and Vaughan, D.J. 2006. Electrical and magnetic properties of sulfides, p. 127-180. In Vaughan, D.J. (ed.), Sulfide Mineralolgy and Geochemistry, Mineralogical Society of America. Chantilly, Virginia. https://doi.org/10.2138/rmg.2006.61.3
Plummer, L.N., Parkhurst, D.L., Fleming, G.W., and Dunkle, S.A. 1988. A computer program incorporating Pitzer's equations for calculation of geochemical reactions in brines. United States Geological Survery Water Investigations Report 88-4153. https://doi.org/10.3133/wri884153
Rieder M., Crelling, J.C., Sustai, O., Drabek, M., Weiss, Z., and Klementova, M. 2007. Arsenic in iron disulfides in a brown coal from the North Bohemian Basin, Czech Republic. International Journal of Coal Geology, 71:115-121. https://doi.org/10.1016/j.coal.2006.07.003
Rimstidt, J.D. and Vaughan, D.J. 2003. Pyrite oxidation: a state-of-the-art assessment of the reaction mechanism. Geochimica et Cosmochimica Acta, 67:873-880. https://doi.org/10.1016/s0016-7037(02)01165-1
Robie, R.A. and Hemingway, B.S. 1995. Thermodynamic properties of minerals and related substances at 298.15 K and 1 bar (10^5 pascals) pressure and at higher temperatures. United States Geological Survey Bulletin, 2131:1-470. https://doi.org/10.3133/b2131
Rosso, K.M., Becker, U., and Hochella, M.F. 1999. The interaction of pyrite (100) surfaces with O-2 and H2O: Fundamental oxidation mechanisms. American Mineralogist, 84:1549-1561. https://doi.org/10.2138/am-1999-1008
Rosso, K.M., Becker, U., and Hochella, M.F. 2000. Surface defects and self-diffusion on pyrite {100}: An ultra-high vacuum scanning tunneling microscopy and theoretical modeling study. American Mineralogist, 85:1428-1436. https://doi.org/10.2138/am-2000-1011
Rosso, K.M. and Becker, U. 2003. Proximity effects on semiconducting mineral surfaces II: Distance dependence of indirect interactions. Geochimica et Cosmochimica Acta, 67:941-953. https://doi.org/10.1016/s0016-7037(02)00990-0
Rosso, K.M. and Vaughan, D.J. 2006a. Reactivity of sulfide mineral surfaces, p. 557-607. In Vaughan, D.J., (ed.) Sulfide Mineralolgy and Geochemistry, Mineralogical Society of America, Chantilly, Virginia. https://doi.org/10.2138/rmg.2006.61.10
Rosso, K.M. and Vaughan, D.J. 2006b. Sulfide mineral surfaces, p. 505-556. In Vaughan, D.J., (ed.) Sulfide Mineralolgy and Geochemistry, Mineralogical Society of America, Chantilly, Virginia. https://doi.org/10.2138/rmg.2006.61.9
Savage, K.S., Stefan, D., and Lehner, S.W. 2008. Impurities and heterogeneity in pyrite: influences on electrical properties and oxidation products. Applied Geochemistry, 23:103-120. https://doi.org/10.1016/j.apgeochem.2007.10.010
Schaufuss, A.G., Nesbitt, H.W., Kartio, I., Laajalehto, K., Bancroft, G.M., and Szargan, R. 1998a. Reactivity of surface chemical states on fractured pyrite. Surface Science, 411:321-328. https://doi.org/10.1016/s0039-6028(98)00355-0
Schaufuss, A.G., Nesbitt, H.W., Kartio, I., Laajalehto, K., Bancroft, G.M., and Szargan, R. 1998b. Incipient oxidation of fractured pyrite surfaces in air. Journal of Electron Spectroscopy and Related Phenomena, 96:69-82. https://doi.org/10.1016/S0368-2048(98)00237-0
Schoonen, M.A.A. and Barnes, H.L. 1991. Reactions forming pyrite and marcasite from solution. 1. Nucleation of FeS2 below 100 degrees C. Geochimica et Cosmochimica Acta, 55:1495-1504. https://doi.org/10.1016/0016-7037(91)90122-l
Schoonen, M.A.A., Elsetinow, A., Borda, M., and Strongia, D. 2000. Effect of temperature and illumination on pyrite oxidation between pH 2 and 6. Geochemical Transactions, 1:23-33. https://doi.org/10.1186/1467-4866-1-23
Schoonen, M.A.A., Harrington, A.D., Laffers, L., and Strongin, D.R. 2010. Role of hydrogen peroxide and hydroxyl radical in pyrite oxidation by molecular oxygen. Geochimica et Cosmochimica Acta, 74:4971-4987. https://doi.org/10.1016/j.gca.2010.05.028
Sippola, H. 2012. Thermodynamic modelling of concentrated sulfuric acid solutions. Calphad-Computer Coupling of Phase Diagrams and Thermochemistry, 38:168-176. https://doi.org/10.1016/j.calphad.2012.06.008
Sklute, E.C., Jensen, H.B., Rogers, A.D., and Reeder, R.J. 2015. Morphological, structural, and spectral characteristics of amorphous iron sulfates. Journal of Geophysical Research-Planets, 120:809-830. https://doi.org/10.1002/2014je004784
Sklute, E.C., Rogers, A.D., Gregerson, J.C., Jensen, H.B., Reeder, R.J., and Dyar, M.D. 2018. Amorphous salts formed from rapid dehydration of multicomponent chloride and ferric sulfate brines: implications for Mars. Icarus, 302:285-295. https://doi.org/10.1016/j.icarus.2017.11.018
Swami, M.S.R. and Prasad, T.P. 1980. Thermal-analysis of iron(II) sulfate heptahydrate in air. 3. Thermal-decomposition of intermediate hydrates. Journal of Thermal Analysis, 19:297-304. https://doi.org/10.1007/bf01915805
Tacker, R.C. 2008a. Reaction of pyrite and clays: experiments in AMD and “pyrite disease". Geological Society of America Abstracts with Programs, 40:24.
Tacker, R.C. 2008b. Influence of clay in pyrite oxidation/hydrolysis and implications for storage. Proceedings of the Sixth International Conference on Mineralogy and Museums, Golden, Colorado, USA, p. 39.
Thomas, P.S., Hirschausen, D., White, R.E., Guerbois, J.P., and Ray, A.S. 2003. Characterisation of the oxidation products of pyrite by thermogravimetric and evolved gas analysis. Journal of Thermal Analysis and Calorimetry, 72:769-776. https://doi.org/10.1023/a:1025001811801
Valsami-Jones, E., Ragnarsdottir, K.V., Putnis, A., Bosbach, D., Kemp, A.J., and Cressey, G. 1998. The dissolution of apatite in the presence of aqueous metal cations at pH 2-7. Chemical Geology, 151:215-233. https://doi.org/10.1016/s0009-2541(98)00081-3
Vaughan, D.J. (ed.). 2006. Sulfide Mineralogy and Geochemistry. Reviews in Mineralogy and Geochemistry, Volume 71. Mineralogical Society of America, Chantilly, Virginia. https://doi.org/10.1515/9781501509490
Vera, M., Schippers, A., and Sand, W. 2013. Progress in bioleaching: fundamentals and mechanisms of bacterial metal sulfide oxidation-part A. Applied Microbiology and Biotechnology, 97:7529-7541. https://doi.org/10.1007/s00253-013-4954-2
Waller, R. 1992. Temperature- and humidity-sensitive mineralogical and petrological specimens, p. 25-50. In Howie, F. (ed.), The Care and Conservation of Geological Materials: Minerals, Rocks, Meteorites and Lunar Finds. Butttesuperrworth-Heinemann, Oxford, U.K. https://doi.org/10.4324/9781315042626
Wang, A., Feldman, W.C., Mellon, M.T., and Zheng, M.P. 2013. The preservation of subsurface sulfates with mid-to-high degree of hydration in equatorial regions on Mars. Icarus, 226:980-991. https://doi.org/10.1016/j.icarus.2013.07.020
Wang, A. and Zhou, Y. 2014. Experimental comparison of the pathways and rates of the dehydration of Al-, Fe-, Mg- and Ca-sulfates under Mars relevant conditions. Icarus, 234:162-173.https://doi.org/10.1016/j.icarus.2014.02.003
Wang, A.A., Ling, Z.C., Freeman, J.J., and Kong, W.G. 2012. Stability field and phase transition pathways of hydrous ferric sulfates in the temperature range 50 degrees C to 5 degrees C: Implication for martian ferric sulfates. Icarus, 218:622-643. https://doi.org/10.1016/j.icarus.2012.01.003
Wang, S.C., Zhao, Y., and Li, S. 2019. Pyrite oxidation inhibition by hydrophobic films for acid mine drainage control at the source. Physicochemical Problems of Mineral Processing, 55:1132-1140. https://doi.org/10.5277/ppmp19035
Wen, X., Liang, Y., Bai, P., Luo, B., Fang, T., Yue, L., An, T., Song, W., and Zhenga, S. 2017. First-principles calculations of the structural, elastic and thermodynamic properties of mackinawite (FeS) and pyrite (FeS2). Physica B, 525:119-126. https://doi.org/10.1016/j.physb.2017.09.007
Xu, W.Q., Tosca, N.J., McLennan, S.M., and Parise, J.B. 2009. Humidity-induced phase transitions of ferric sulfate minerals studied by in situ and ex situ X-ray diffraction. American Mineralogist, 94:1629-1637. https://doi.org/10.2138/am.2009.3182
Yoder, C., Pasteris, J., Worcester, K., Schermerhorn, D., Sternlieb, M., Goldenberg, J., and Wilt, Z. 2012a. Dehydration and rehydration of carbonated fluor- and hydroxylapatite. Minerals, 2:100-117. https://doi.org/10.3390/min2020100
Yoder, C.H., Pasteris, J.D., Worcester, K.N., and Schermerhorn, D.V. 2012b. Structural water in carbonated hydroxylapatite and fluorapatite: confirmation by solid state H-2 NMR. Calcified Tissue International, 90:60-67. https://doi.org/10.1007/s00223-011-9542-9
Young, E.J., Myers, A.T., Munson, E.L., and Conklin, N.M. 1969. Mineralogy and geochemistry of fluorapatite from Cerro de Mercado, Durango, Mexico. United Stataes Geological Survey Professional Paper, 650-D:D85-D92.
Zhang, X.L., Kou, J., and Sun, C.B. 2019. A comparative study of the thermal decomposition of pyrite under microwave and conventional heating with different temperatures. Journal of Analytical and Applied Pyrolysis, 138:41-53. https://doi.org/10.1016/j.jaap.2018.12.002
